INTRODUCTION
The study of the molecular epidemiology of parasitic infections and their vectors is meant to answer the same kinds of questions as those of bacterial or viral infections. As with bacteria, the molecular epidemiology of eukaryotic infections follows the distribution and dynamics of microbial DNA. The key difference, however, is precisely this biology, which defines a distinct approach to molecular epidemiologic investigation of infections caused by eukaryotic organisms. In bacterial reproduction, each individual passes down an identical copy of all the DNA to the next generation. Some eukaryotic pathogens behave reproductively in similar ways to bacteria and reproduce asexually, while others have sexual reproduction for at least part of their life-cycle. The individual is able to generate a clone of itself by binary fission to produce two identical organisms, and if successful, will produce large numbers to the detriment of its host. Asexually reproducing organisms can also exhibit promiscuous horizontal gene transfer, which can be a major source of variation and adaptation (19), but this is not sex. Sex is the biologically necessary programmed recombination (crossing over) and random shuffling (reassortment) of chromosomal DNA in the process of reproduction. This results in an enormous reservoir of variation. Bacteria in nature are heterogenous conglomerates or communities (13, 19), but when they cause disease, especially in epidemics, it is generally a clone that is responsible and that we track (Chapter 2). Sexual reproduction in some protozoa, many parasitic worms and most vectors, however, never results in a clone with the exception of identical twins. There is genetic conservation, however, within a group of organisms that tends to breed together. In genetics, this is the working definition of a population. For sexually reproducing organisms, the population is the epidemiologic unit to track. Within the group, allele frequencies and thus traits are conserved under well-defined conditions. The unique power of the genetics of populations is that it reflects not only present individuals but also the population’s past and the future potential for subsequent generations (5). Many parasites exhibit both sexual and asexual modes of reproduction, but these life stages are distributed in different hosts. Treatment of their molecular epidemiology is doubly complex, but can be simplified for some questions by considering their biology just in the human host. The whole field of population genetics is perhaps the most complex area of genetics, but it arises from simple precepts. This chapter will outline the basic models used in population genetics and are directly applicable to problems of public health epidemiology.
KEY POINTS
DEFINING GENOTYPE IN EUKARYOTIC ORGANISM
Some terms may not be familiar to some readers, so it is important to define these early. One of the dividing lines between bacteria and sexually reproducing parasites and vectors of human disease is their physical structure and organization. Sexually reproducing organisms will pass some portion of their life cycle where their chromosomes (Figure 5.1) exist as nearly identical pairs (diploid). Some organisms, malaria in particular, also have only one copy (haploid) during their asexual stage, and this is the stage that infects humans. A similar location on each of the chormosomes is a locus, and differences between loci are alleles.

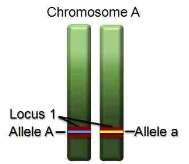
The geometry of DNA also strongly differentiates bacteria from eukaryotes (Figure 5.2). Prokaryotes have a single[1], circular chromosome whereas even the simplest eukaryotes, yeast, have at least 16 linear chromosomes. A specific marker on a bacterial chromosome will always be transmitted at reproduction together with any other marker or trait. The same also occurs with an asexually reproducing eukaryote despite having multiple linear chromosomes. A marker on the genome of a sexually reproducing eukaryote, by contrast, will have a 50% chance of being transmitted away from any marker it is not very close to. The labeling of each allele present at the same locus on each chromosome constitutes the genotype. A locus with the same polymorphism at the same site on each of the chromosome is homozygous, and with a different polymorphism is heterozygous.





 Figure 5.2
Figure 5.2
OPTIONS FOR MOLECULAR EPIDEMIOLOGY OF EUKARYOTES
HARDY-WEINBERG EQUILIBRIUM: THE POPULATION NULL HYPOTHESIS
Populations have a mathematical definition based on allele frequencies, which ultimately contributes to the development of tools for key measures of differentiation and diversity. Allele frequencies can differentiate populations, and genotypic frequencies can do so with even greater resolution. The relationship between allelic frequency and genotypic frequency has a simple mathematical relationship which is the definition of a population. If we use the letters “A” and “a” to represent different alleles at a single diallelic locus and “p” and “q” to represent their respective frequencies, a population with p=0.8 and q=0.2 is clearly different from a population where p=0.2, q=0.8, especially where this kind of result is found at multiple loci. Allele frequencies are not always the most sensitive measure of differentiation. The same allele frequency may still be found in what are clearly distinct populations if assessed for genotypic frequencies. Alleles combine to form genotypes, so the genotypic frequency is a function of the allelic frequencies. For a diallelic locus where we know the frequency of each allele, the sum of these frequencies is 1 or (p + q = 1). For sexually reproducing organisms the next generation arises from the combination of alleles from a pool of males with alleles from a pool of females. If we imagine that individuals from these pools will pair at random, the subsequent distribution of alleles in genotypes is equivalent to rolling a pair of dice. For independent, random events the probability of 2 events occurring simultaneously is the product of their frequencies [(p + q)female • (p + q)male = 1]. The genotypic frequencies of the offspring for such a population should be p2 + 2pq + q2, if all assumptions are met, where p2 and q2 are the frequencies for the homozygotes and 2pq the heterozygotes. This is the well-known Hardy-Weinberg equilibrium (HWE). This simple quadratic equation is the basis for all population genetics even when it is not measured directly. It represents the expected genotypic frequencies from a given set of allelic frequencies. It is one of the most stable mathematical relationships in nature. It is so much the expectation that when not observed in sequencing projects, it can suggest sequencing errors. It is the null hypothesis and mathematical definition of a stable population. The relationship HWE describes is true under a set of 5-10 assumptions that represent the most important factors that influence population genetic structure. The 5 most common assumptions are that there is:
1) Random mating (panmixia, assortative mating)
2) No selection
3) No migration
4) An infinite population
5) No mutation
It is rare to have any of these assumptions met in nature, but the proportions are so resilient that the assumptions have to be severely violated to disturb this relationship, and even so, the proportions will be reestablished within 1-2 generations once the population is stabilized. As with most models, the underlying assumptions are the most important aspects. They are the basis for most conversations in population genetics.
MARKERS
Microsatellites, single nucleotide polymorphisms (SNPs) and sequencing are currently the genetic elements most employed in population genetics. Microsatellites are short tandem repeats of 2-8 nucleotides (reviewed in [Ellegren, 2004 #128]).  Microsatellites have fallen out of favor in studies of statistical genetics or gene finding, since SNPs and sequencing provide better resolution at the level of individuals. Microsatellites, however, remain important in population genetics since they are mostly neutral for selection and have higher allelic richness and information content. Their rapid mutation rate (10-2 ─ 10-5 per generation) and step-wise mode of mutation can limit their application to questions that extend over short time scales and to certain statistical approaches. SNPs have lower rates of mutation (10-8) in eukaryotes, often are diallelic, are ten times more abundant (10, 22) and have high processivity and scorability. Sequencing essentially provides a very dense panel of SNPs and identifies rare variants as well as structural polymorphisms. Mitochondrial and ribosomal DNA markers are much less abundant, less polymorphic and thus less informative than microsatellites or SNPs. Some are under selection and in the case of mitochondrial DNA, the genome is haploid (only 1 copy of chromosomal DNA) and may or may not have sex-specific inheritance depending on the species. They are useful for phylogeny studies, may be more economical to use in laboratories with limited capabilities and are sometimes combined with other markers.
MEASURES OF DIFFERENTIATION AND DIVERSITY
Areas most often addressed using population genetics are evolution and conservation. These two areas deal with essentially the same phenomenon, but at different time scales, thus the questions, the approaches and the interpretation will differ depending on the nature of the problem. The relevant public health questions in population genetics focus on identity and dynamics of the group rather than individuals over short time scales and directed at the control or extinction of a parasite or vector. Who’s sleeping with whom, modes of reproduction, evolution or the last common ancestor are all important in different contexts. They may be useful to help explain anomalies and can influence interpretation, but they are rarely answers to issues of control or intervention. Understanding how diverse a population is or the degree of difference between populations combined with good study design will contribute directly to determining the impact of control measures, host or parasite demographics, resistance, risk and resilience or fragility of the population.
The field of population genetics depends heavily on mathematical analyses, some simple and some very sophisticated, to answer these questions. Mathematical treatments of all of the indices and statistics of differentiation and diversity can be easily obtained from textbooks or publications and will not be included here. Fortunately for the mathematically challenged, many open source, individual computer programs are available as well as modules in R. The risk that goes with all readymade programs is a failure to understand what is being asked or the assumptions and limitations of the approach being taken. A list of some frequently used programs is provided at the end of this chapter (Table 5.1).
POPULATIN DIFFERENTIATION
FST, GST, G’ST: In addition to the Hardy-Weinberg equilibrium, populations can be further differentiated by other statistical tests. This is a family of statistics developed as the fixation index (FST) in the 1950’s by Sewell Wright and Gustave Malécot to describe the likelihood of homozygosity (“fixation” in the terminology of the time) at a single diallelic locus based on heterozygosity of a subpopulation compared to the total population. Theoretically, values should range from 1 (no similarity-every individual is genotypically different) to 0 (identity-every individual is genotypically same). Nei (16) extended the FST to handle the case of more than two alleles and developed the GST[LR1]. Although the term “FST” is often used in the literature, formally most studies today will employ the GST. When highly polymorphic loci, such as microsatellites, are genotyped, the GST severely underestimates differentiation and will not range from 0 – 1. Hedrick (11) adjusted the range of values for the GST by dividing it by its maximum possible value given the markers used. This is the G’ST. The G’ST makes possible the full range of differentiation. FST and GST relate to inherent properties of populations and contain evolutionary information lost by the G’ST transformation. FST-like measures have been and continue to be widely used to describe population structure, and their characteristics and behavior are well-known. There are additional related statistics (e. g. φST (4), AMOVA, RST (20), θ (25)), that address other aspects of differing genetic models, unequal sample sizes, accounting for haploid genomes, mechanism of mutation and selection.
D. [LR2]This is sometimes referred to as Jost’s D, since there are numerous other D’s related to genetics and statistics. There can be logical inconsistencies for estimating differentiation based directly on heterozygosity. Ratios of pooled subpopulation to total population diversities tend toward zero when the subpopulation diversity is high (12). Jost’s D is based on the effective allele number (see below). Unlike those based directly on heterozygosity, it has the property of yielding a linear response to changes in allele frequencies and is independent of subpopulation diversity. Unlike FST, G’ST and similar indices, Jost’s D does not carry information relevant to the evolutionary processes responsible for the present composition of a subpopulation. It is described by supporters and detractors as purely a measure of differentiation (26). It was never meant to do more.
Whitlock provides one of the best comparisons of these 2 approaches:
This (Jost’s) D differs from FST in a fundamental definitional way: FST measures deviations from panmixia[2], while D measures deviations from total differentiation. As a result, their denominators differ, and thus, the two indices can behave quite differently. D indicates the proportion of allelic diversity that lies among populations, while FST is proportional to the variance of allele frequency among populations. D is more related to the genetic distance between populations than to the variance in allele frequencies; it may be preferable to call D a genetic distance measure (26).
There has been controversy about the use of these different types of indices. There should not be. They clearly address different questions and resolve different analytic problems. It should be recognized that the G’ST and Jost’s D yield fairly similar results when the number of populations is small and the markers have a small number of alleles. The G’ST and Jost’s D have given similar results in our own studies using microsatellites (2) and in simulation (26) with G’ST values slightly higher than those of Jost’s D. Some authors recommend calculating both GST and Jost’s D, in part to satisfy everyone and in part to obtain the useful information about population diversity their departure may provide.
In relation to public health, most questions about parasites and vectors deal with near term events of <10-20 generations rather than events on an evolutionary scale. The result we often seek is simply to know how different the allelic composition of one group is compared to another using microsatellite markers. In addition, the population or subpopulation structures may be such that there cannot be strong assumptions of HWE. We generally have used D for questions subpopulation differentiation. The use of other markers, such as SNPs and sequencing may require other approaches such as FST or principal component analysis (8). Apart from differentiation, individuals can be clustered into populations by model-based, structured association methods used in programs such as Structure (18) and Admixture (1). These programs use approximation to HWE as part of their clustering rule.
DIVERSITY
Diversity like differentiation has myriad formulations and interpretations. The simplest expression is mean heterozygosity (H). For microsatellite data this is usually high due to the intrinsically high mutation rate of these markers, and markers with higher variability are usually selected. Allelic richness (Ar) is simply a count of the number of alleles at each locus. Differences in sample size will necessarily result in differences in allele number. This is usually adjusted for by statistical methods such as rarefaction (15) to standardize sample sizes between comparison groups. The effective allele number (Ae) is also a measure of diversity, but is already adjusted for sample size. It represents the number of alleles with equal frequencies that will produce the same heterozygosity as that of the target population.
The most informative measure of diversity is the effective population size (Ne), a concept also introduced by Sewell Wright. It is designed to address the essential reason that diversity is important, namely, it reflects the strength of genetic drift. Genetic drift is the effect of random transmission of alleles during reproduction to succeeding generations. When numbers of reproducing individuals are small, the genetic composition of the population of offspring can differ by chance from what is expected given the composition of the parents. If two coins are flipped, it would not be that unusual for both to come up heads. If a thousand coins are flipped, the ratio of heads to tails will always be very near the expected 50:50 ratio. Genetic drift is stronger when populations are small or reduced, and weakens the strength of adaptive selection.
Like differentiation, there are several formulations for Ne that can provide different values and are designed to measure different aspects of the population. The breeding Ne is the probability of identity by descent for two alleles chosen at random. It is a retrospective assessment of population diversity. The variance Ne assesses the variance of the offsprings’ allele frequency, and is thus forward looking. It measures recent population changes that affect its genetic composition. Ne can represent the number of actively breeding individuals in the population or the number of individuals in an ideal population needed to reconstitute the diversity in an actual population. It is almost always less than the census population (Nc). It is a key value in conservation genetics and population genetics in general, since it reflects the history and future potential of a population. Increasing drift (decreasing Ne) tends to neutralize the force of directional selection, permits retention of deleterious mutations and hampers the ability of populations to adapt to stresses (9).
Despite its importance, Ne can be difficult to estimate in wild populations due to uncertainties of the demographic, genetic and biological context (17, 24). It can be affected by sample size, overlapping generations, sampling interval, sex ratios, gene flow, age-structure, variation in family size, fluctuating population size or selection. Increasing the numbers of markers is less important than large samples for accurate estimates; as much as 10% of the Ne has been recommended [Palstra, 2008 #84]. Its interpretation can also be uncertain. Estimated Ne has been used as an aid in predicting extinction using the concept of a minimum viable population size. Some have suggested that at an Ne of 50-500 a population will experience extinction in the short- or long-run (7). Others have argued that this might occur at Ne = 5000 (14). While it is clear that lack of diversity has an impact on extinction (21), it is also clear that there cannot be a universally accepted number for the minimum viable population size (6, 23). In any case, theory suggests that there is a number defined by the amount of genetic drift below which populations are likely to go extinct on their own. The range for this number is context-specific and will require multiple species-specific studies under multiple conditions. This kind of analysis might contribute to developing a stopping rule as control measures approach elimination.
[1] Leptospira spp. are an exception with 2 circular chromosomes.
[2] The condition where all individuals have an equal opportunity to reproduce with all other individuals
[LR1]G–?
[LR2]Does it stand for something?
You have to be 100% sure of the quality of your product to give a money-back guarantee. This describes us perfectly. Make sure that this guarantee is totally transparent.
Read moreEach paper is composed from scratch, according to your instructions. It is then checked by our plagiarism-detection software. There is no gap where plagiarism could squeeze in.
Read moreThanks to our free revisions, there is no way for you to be unsatisfied. We will work on your paper until you are completely happy with the result.
Read moreYour email is safe, as we store it according to international data protection rules. Your bank details are secure, as we use only reliable payment systems.
Read moreBy sending us your money, you buy the service we provide. Check out our terms and conditions if you prefer business talks to be laid out in official language.
Read more