1. (10) We learned in Woong Song’s seminar ( S W. Huskey, C Zhu, A Fredenhagen, J Kühnöl, A Luneau, et.al. KAE609 (Cipargamin), a New Spiroindolone Agent for the Treatment of Malaria: Evaluation of the Absorption, Distribution, Metabolism, and Excretion of a Single Oral 300‐mg Dose of [14C]KAE609 in Healthy Male Subjects. Drug Metabolism and Disposition May 2016, 44 (5) 672‐682) that Cipargamin had an elimination t1/2 of about 33 hours, which is not good for marketed drugs.
a. Based on your drug metabolism experience, can you explain why cipargamin has such a long half-life.
KAE609 [(1′R,3′S)-5,7′-dichloro-6′-fluoro-3′-methyl-2′,3′,4′,9′-tetrahydrospiro[indoline-3,1′-pyridol[3,4-b]indol]-2-one] is a potent, fast-acting, schizonticidal agent being developed for the treatment of malaria. KAE609 showed low clearance, moderate volume distribution, and a long t1/2 in rats and dogs.
Cipargamin undergoes an unusual C-C bond cleavage, followed by a 1,2-acyl shift to form a ring expansion product M37, that is showed in figure 1. This isomerization and rearrangement reaction was catalyzed by CYP3A4. The mechanism by which KAE609 undergoes oxidation includes a single electron transfer to form a radical cation, followed by C-C bond cleavage to form an isocyanate radical cation intermediate. The isocyanate radical cation is reduced by a single electron transfer, followed by protonation. Nucleophilic attack by the amine on the carbonyl carbon of the isocyanate leads to ring closure to form M37[1]. However, this mechanism tents to be slow causing a delay in the excretion of this drug, as a consequence its half-life would be very long.
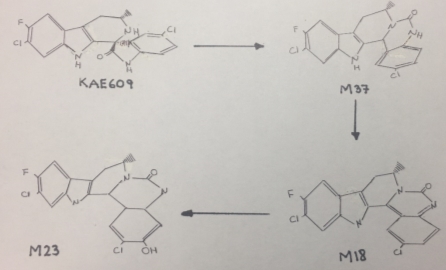 It is important to remark that the chemical structure of Cipargamin contains many deactivating substituents that withdraw the electrons making this compound difficult to oxidize and to eliminate.
It is important to remark that the chemical structure of Cipargamin contains many deactivating substituents that withdraw the electrons making this compound difficult to oxidize and to eliminate.
b. As a member of the drug development team for this antimalarial drug, what structural changes to this drug would you suggest, based on your drug metabolism experience, that would shorten the elimination t1/2 to a more reasonable time.
This molecule presents aromatic deactivators as two molecules of chlorine and two fluorine. Halogens are electron withdrawing groups leading to destabilize the intermediate cations, therefore the hydroxylation of the two benzene rings is inhibited. The carbonyl group cannot the oxidized due the steric hindrance and it also plays a role as deactivating substituent. Therefore, I would suggest introduce electron-donating or activating substituents as hydroxyl group or an alkyl chain instead of the electron-withdrawing groups. The more electron rich the aromatic ring, the faster the aromatic hydroxylation takes places and the lower the half-life facilitating the elimination of this drug.
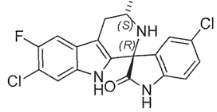
Figure 2. Chemical Structure of KAE609. D. Williams
2. (10) a. Explain what a P450 carbon‐centered oxidation is and Include the proposed mechanisms for P450 carbon hydroxylation.
A P450 carbon-centered oxidation is the reaction by which a radical is generated by the abstraction of a hydrogen atom or an electron from a substrate. Then, the carbon centered radical reacts with the ferric-bound hydroxyl compound to produce an oxidized product. There are two proposed mechanism for P450 carbon hydroxylation:
b. Explain the mechanism of oxygen insertion for P450?
The CYP 450 familiy enzymes are also known as monooxygenases or mixed-function oxidases, due to its ability to incorporate one atom of molecular oxygen into a substrate. The hydroxylation catalyzed by this enzyme proceed by hydrogen abstraction followed by a reaction of carbon-centered radical with an iron-bound hydroxyl radical in a process termed oxygen rebound. In order to insert an oxygen atom into an organic substrate molecule, these enzymes activate dioxygen concomitant with the reduction of the other atom of oxygen to water. The mechanism is as follows:
The mechanism of oxygen insertion begins when the oxidized P450 complex (Fe+3-P450) binds reversibly with a substrate that is going to be hydrolyzed, forming a complex that displaces a molecule of water. Then, NADPH-cytochrome P450 reductase (a CYP redox partner) provides an electron to reduce the P450 heme to ferrous state.
A molecular oxygen binds to the reduced enzyme-substrate complex as the sixth ligand to form a nucleophilic complex (enzyme-O2-substrate) that it is rearranged by resonance due to the strong electronegativity of oxygen to form a Fe+3-P450*RH-superoxide anion O2-1 complex. Then, there is a transfer of a second electron from the cytochrome b5 (another CYP redox partner) that reduces the complex previously formed in an unstable peroxide complex. After that, there is a protonation that produce a cleavage of the O-O bond releasing water and forming a highly electrophilic porphyrin-radical cation intermediate: ferryc oxenoid, called Compound I, which is the most favorable and stable oxygen cysteine porphyrin radical complex (Fe+4=O).
The ferryc oxenoid species abstracts a hydrogen atom from the substrate, producing a carbon radical intermediate, that undergoes a recombination with the ferric-bound hydroxyl radical, called Compound II (Fe+4=OH), where the retained oxygen atom is inserted into the substrate. Finally the oxidized product is released and the heme iron returns to the ferric state[4].
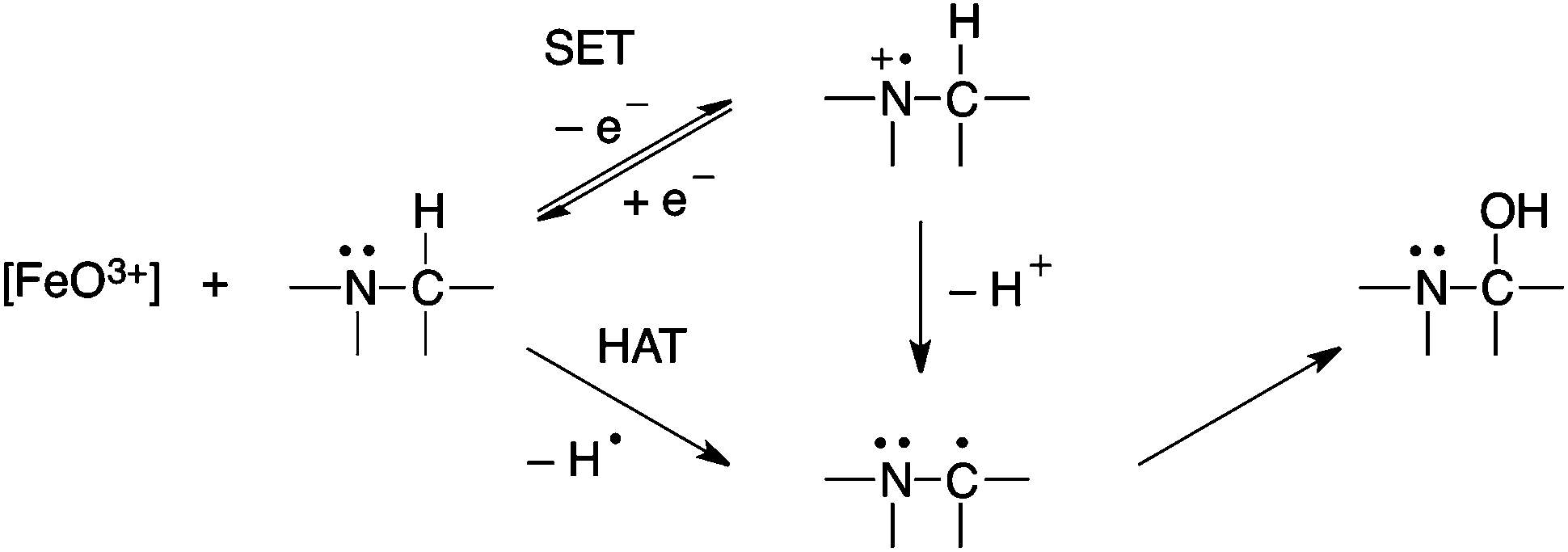
Figure 3. Example of HAT and SET mechanism.[5]
3. (10) In Cullen’s seminar (JK. Towles, RN. Clark, M D. Wahlin, VUttamsingh, et.al. Cytochrome P450 3A4 and CYP3A5‐Catalyzed Bioactivation of Lapatinib. Drug Metabolism and Disposition October 2016, 44 (10) 1584‐1597), what was the purpose of incubating lapatinib with the following inhibitors: α‐naphthoflavone, ticlopidine, sulfaphenazole, quinidine,4‐methylpyrazole, and ketoconazole.
Lapatinib is an orally administered dual inhibitor of tyrosine kinases epidermal growth factor receptor 1 and human epidermal growth factor receptor 2 used in the treatment of breast cancer. This drug is primarily bioactivated by CYP3A4 and CYP3A5 in a reactive quinoneimine metabolite that induces hepatotoxicity. This metabolite may covalently modify cellular proteins leading to cell stress and/or immune activation. Additionally, this quinoneimine can be conjugated to form a quinoneimine-GSH adducts
In order to identify the role of each CYP450 isoform involved in the bioactivation of lapatinib, the authors of this study incubated lapatinib in the presence of different P450-selective chemical inhibitors in human liver microsomes. The inhibitors used to block the different CYP450 isoforms were: α‐naphthoflavone (CYP1A2 inhibitor), ticlopidine (CYP2B6 and CYP2C19 inhibitor), sulfaphenazole (CYP2C9 inhibitor), quinidine,4‐o(SR-9186 and CYP3cide). The purpose of these substances is inhibit a specific CYP450 enzyme to quantify the levels of lapatinib metabolites and GSH adducts in each sample and then compare it with a control that does not contain any inhibitors. In other words, if the concentration of those metabolites are lower than the control we can suggest that the inhibition of that specific CYP450 isoform is blocking the bioactivation of the drug. Those results will demonstrate which isoforms are involved in the bioactivation of lapatinib.
In this study, it was found that ketoconazole, CYP3A4/5 inhibitor, reduce the formation of the reactive quinoneimine and its GSH adducts by 89% compared with the control confirming that these enzymes are primarily responsible for the production of the toxic metabolite. There is also a reduction of the GSH adduct formation from the O-dealkylated lapatinib metabolite with the CYP2C9 inhibitor, sulfaphenazole, demonstrating that CYP2C9 is also involved in the activation of lapatinib. In addition, the CYP3A4-selective inhibitors SR-9186 and CYP3cide decreased quinoneimine-GSH adduct formation to a lesser extent than the pan-CYP3A inhibitor ketoconazole. This finding indicates that the remaining CYP3A5 activity in human liver microsomes is likely responsible for generating the reactive quinoneimine-GSH adducts from lapatinib.
4. (10) a. Why doesn’t Vmax change with competitive inhibition, but Km doesn’t change, remains constant. Please explain.
A competitive inhibitor is any compound which closely resembles the chemical structure and molecular geometry of the substrate. The inhibitor competes for the same active site as the substrate molecule and it may interact with the enzyme at the active site, but no reaction takes place. In other words, the enzyme may be bound to the inhibitor or the substrate, but it cannot bind both at the same time. The inhibitor is trapped on the enzyme and prevents any substrate molecules from reacting with the enzyme. However, a competitive inhibition is usually reversible if sufficient substrate molecules are available to ultimately displace the inhibitor. Therefore, the amount of enzyme inhibition depends upon the inhibitor concentration, substrate concentration, and the relative affinities of the inhibitor and substrate for the active site.
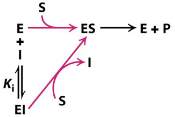
Figure 4. Scheme of a competitive inhibition reaction
In a competitive inhibition, the maximum velocity (Vmax) of the reaction is unchanged, while the apparent affinity of the substrate to the binding site is decreased. Any competitive inhibitor concentration can be overcome by increasing the substrate concentration in which case the substrate will outcompete the inhibitor in binding to the enzyme and dissociate the enzyme-inhibitor complex. This means that increasing concentrations of substrate will decrease the chance of inhibitor binding to the enzyme.
In a competitive inhibition the true values of the Km do not change, because the true affinity of the substrate for the enzyme is not going to change. The measured values of Km in the presence of the inhibitor are altered, and are called the apparent Km. The apparent Km varies depending on the inhibitor concentration involved.
The apparent Vmax is unaffected by a competitive inhibitor. This is because once substrate binds, the reaction proceeds normally, and therefore Vmax depends only on the maximum possible ES complex concentration (and on the kcat, but kcat is unaffected by a competitive inhibitor), and the maximum ES concentration depends only on the total amount of enzyme present. In other words, if the substrate concentration is high enough the enzyme will reach the same Vmax as without the inhibitor. In contrast, the apparent Km increases as a result of the inhibitor because it raise the concentration of substrate required for a given velocity and it will require a higher concentration of substrate to achieve this, that is why the apparent Km of the enzyme will be higher.
The next figure is the Lineweaver-Burk double reciprocal plot that shows a series of lines crossing the y (1/v) axis at the same point means that Vmax is unchanged, but with a decreasing value of 1/Km (and hence a higher Km) in the presence of the competitive inhibitor:
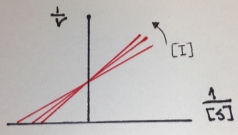
Figure 5. Lineweaver-Burk double reciprocal plot of a competitive inhibition
b. Why are the reciprocal plots very useful for detecting Michaelis‐Menten and non‐classical Michaelis‐Menten inhibition behavior?
In a Michaelis-Menten plot is difficult to obtain an accurate value of Km, which is the concentration of the substrate at half of the maximum velocity (Vmax), because Vmax is approached asymptotically. However, Vmax can be accurately determined if the Michaelis-Menten equation is transformed into one that gives a straight-line plot given by the reciprocals of Michaelis-Menten equation as the Lineweaver–Burk plot, Eadie-Hofstee plot or Hanes-Woolf plot.
In the case of Michaelis-Menten inhibition behavior, the Lineweaver–Burk plot was widely used to determine important terms in enzyme kinetics, such as Km and Vmax. The y-intercept of the graph is equivalent to the inverse of Vmax and the x-intercept of the plot represents −1/Km. This plot is useful for distinguishing between competitive and noncompetitive inhibitors. In competitive inhibition, the intercept on the y-axis of the plot of 1/V0 versus 1/[S] is the same in the presence and in the absence of inhibitor, although the slope is increased. Since Vmax is unaffected by competitive inhibitors, the inverse of Vmax also doesn’t change. However the apparent Km increases as a result of the inhibitor because it raise the concentration of substrate required to achieve a given velocity.
On the other hand, in a noncompetitive inhibition the inhibitor can combine with either the enzyme or the enzyme-substrate complex. The value of Vmax is decreased to a new value called apparent Vmax, and so the intercept on the vertical axis is increased. In contrast with Vmax, Km is not affected by a noncompetitive inhibition.
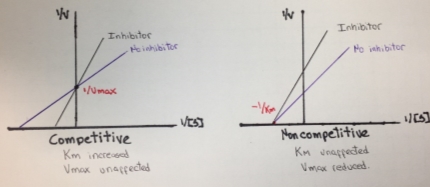
Figure 6. Lineweaver-Burk plot for competitive and noncompetitive inhibition.
Additionally, the reciprocal plots are useful for detecting non-Michaelis-Menten behavior as multiple enzymes catalyzing the formation of the same product, allosteric behavior or non-specific binding of substrates to protein or lipid. The enzymes that follows this kind of behavior do not demonstrate hyperbolic saturation kinetics, or typical Michaelis-Menten kinetics and in graphs of initial velocity vs substrate demonstrate sigmoidal dependency of v on S.
The Eadie-Hofstee graph plot v against v/S will hence yield Vmax as the y-intercept, Vmax/Km as the x-intercept, and Km as the negative slope. This graph also linearizes the Michaelis-Menten equation and it is useful to identify an enzyme that does not follows Michaelis-Menten kinetics, because the enzyme shows a clear deviation from linearity. In the next two figures, some typical and atypical kinetics plots are shown:
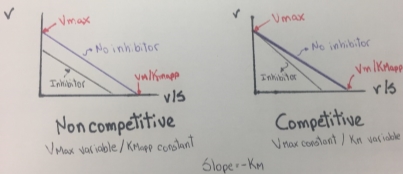
Figure 7. Eadie- Hofstee plots obtained from Competitive and noncompetitive inhibition
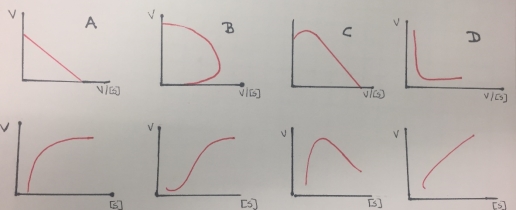
Figure 8. Eadie- Hofstee (first row) and Michaelis-Menten (second row) kinetics plots obtained from enzymes that do not follow a Michaelis-Menten inhibition behavior(except A). A represents a single enzyme, B represent a sigmoidal activation (allosteric effect), C is related to partial inhibition and D represents a biphasic kinetic profile.
5. (10) a. Explain why the enzyme kinetics of mechanism‐based inhibition resembles non‐competitive inhibition and not reversible inhibition, and how can you distinguish mechanism based inhibition from non‐competitive inhibition.
A mechanism-based inhibition is a type of inhibition in which a relatively unreactive compound has a structurally similarity to a given substrate and inhibits it binding to the target enzyme which produces a reactive group. This reactive group reacts to form a covalent bond during the catalytic cycle to form an irreversible enzyme-inhibitor complex.
This type of enzyme inhibition results in the stoichiometric covalent modification of a side chain on an amino acid in the active site of an enzyme. The inhibitor chemically resembles a substrate and binds in the active site in the same way as the substrate binds. The inhibitor, however, has a functional group, that is replaced by a nucleophile in the enzyme active site. This covalent enzyme-inhibitor complex forms irreversibly complex reversibly inactivating the enzyme, resembling a non-reversible inhibition. Therefore this type of inhibition is called “suicide inhibition” or affinity labeling and the inhibitor is called a “suicide inhibitor”. The suicide inhibitor inactive irreversible an enzyme and reduces the formation of enzyme-substrate complex. The Vmax value is reduced and the inhibition cannot be overcome by adding more concentration of the substrate, thereby the km remains constant. In this regard, mechanism-based inhibition resembles non-competitive inhibition. In the next figure, it can be illustrated a Michaelis-Menten and a Lineweaver-Burk plots of a mechanism-based inhibition that resembles a noncompetitive inhibition behavior that was showed in figure 6.
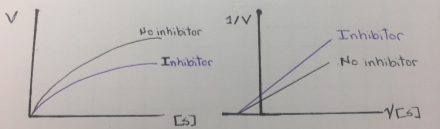
Figure 9. Lineweaver-Burk plot of mechanism-based inhibition.
It can be distinguished from a reversible inhibition because the original enzyme is no longer detectable after the inhibition process and it is necessary synthesized new enzyme molecules to continue the enzyme activity. Also it can be distinguished from noncompetitive inhibition by investigating the time-dependence of the inhibition process, because the suicide inhibitors often act as time-dependent inhibitors, while reversible inhibitors, as noncompetitive, do not show time-dependence. Additionally, for irreversible inhibitors, the IC50 value changes over the time, but in the case of a reversible inhibitors, the IC50 remains independent of the time.
The simplest method to differentiate a noncompetitive inhibition from a mechanism-based inhibition is eliminating the excess of inhibitor by dialysis or gel filtration. The activity of the enzyme will return in the case of noncompetitive inhibition, but in the case of mechanism-based inhibition the activity is not recovered because the suicide inhibitor inactivates the enzyme.
Another method is plot Vmax against total enzyme concentration. An enzyme in presence of a noncompetitive inhibitor will generate a linear plot that starts from the origin. However, its plot will show a lower slope than the plot obtained from the enzyme without inhibitor. On the other hand, the plot obtained in the presence of a suicide inhibitor (in increasing concentrations of the enzyme) will start at a certain distance from the origin on ‘X’ axis, in a point where there is no more enzyme bound to the suicide inhibitor. This plot would be parallel to the control curve and the distance from zero to the start of this plot on ‘X’ axis will yield the concentration of that suicide inhibitor.
b. Explain the statement that all noncompetitive inhibition is allosteric inhibition, but not all allosteric inhibition is noncompetitive inhibition.
Allosterism is the regulation of an enzyme by binding an effector molecule at a site other than the enzyme’s active site. The site to which the effector binds is called the allosteric site. Allosteric sites allow effectors to bind to the protein resulting in a conformational change. Effectors that enhance the protein activity are referred as allosteric activators, whereas those that decrease the protein activity are called allosteric inhibitors.
An allosteric inhibition is promoted by a substance that binds to an enzyme inducing the inactive form of that enzyme. This inhibitor produce a conformational change of the enzyme altering the shape of the active site and changing the enzyme catalytic activity.
On the other hand, a noncompetitive inhibitor is defined as a substance that inhibits the action of an enzyme by binding to the enzyme at a location other than the active site. The inhibitor does not prevent the substrate from binding to the active site, but it still prevents the reaction from completing. A noncompetitive inhibitor binds at an allosteric site, therefore all noncompetitive inhibitions is a type of allosteric inhibition. However, not all allosteric inhibitions are noncompetitive, due to the fact that certain forms of allosteric inhibition can prevent the substrate from binding to the active site, therefore, allosteric inhibition can be noncompetitive or competitive. In other words, noncompetitive inhibitors completely prevent a reaction by combining with non-catalytic sites from happening while allosteric competitive inhibitors reduce the enzyme’s affinity for its substrate.
Figure 10 is showing a noncompetitive inhibition, where the inhibitor binds an allosteric site. In contrast to figure 11, that is illustrating a competitive inhibition where the inhibitor binding to the allosteric site stopping the substrate binding.
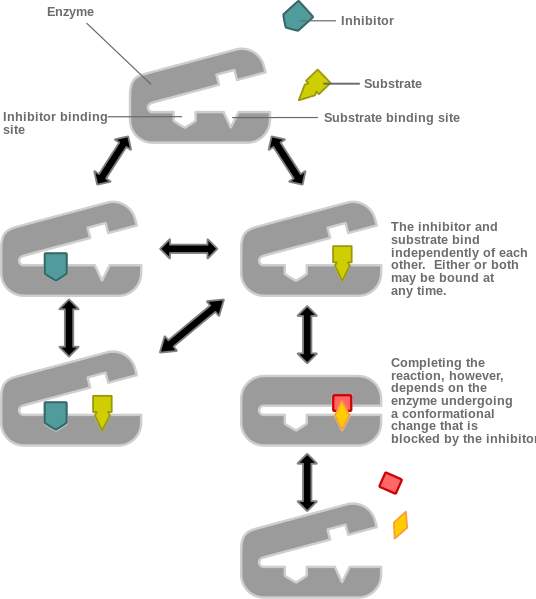
Figure 10. Noncompetitive allosteric inhibition.[6]
![[​IMG]](https://images.ukdissertations.com/17/0020608.011.jpg)
Figure 11. Competitive allosteric inhibition.[6]
6. (10) In Enas’s seminar, (Inzhong Liu, Peter J. Flockhart, Deshun Lu, Wei Lv, Wenjie Jessie Lu, Xu Han, Mark Cushman, and David A. Flockhart, Inhibition of Cytochrome P450 Enzymes by the E‐ and Z‐Isomers of Norendoxifen, Drug Metab Dispos 2013 41:1715‐1720), how did the authors come to the conclusion that the minor metabolite of tamoxifen, norendoxifen is the most potent aromatase inhibitor among the known metabolites that they have tested.
Tamoxifen is a drug used to prevent breast cancer in women and treat breast cancer in women and men. It is a selective estrogen receptor modulator that reduces the estrogenic effects by antagonism of estrogen binding to the estrogen. It has a complex metabolic profile involving both active and inactive metabolites receptors. There are three active metabolites of this drug: N-desmethyltamoxifen (N-DMT), 4-hydroxy-N-desmethyltamoxifen (endoxifen) and N,N-didesmethyl-4-hydroxy-tamoxifen (norendoxifen), which is the minor metabolite. Unlike tamoxifen, norendoxifen is not a selective estrogen receptor modulator (SERM), and instead has been found to act as a potent and selective competitive inhibitor of aromatase. Drugs with dual SERM and aromatase inhibition activity may have therapeutic potential as antiestrogens in the treatment of estrogen receptor-positive breast cancer. The aromatase, also called estrogen synthetase or estrogen synthase, is an enzyme responsible for a key step in the biosynthesis of estrogens. It is CYP19A1, a member of the cytochrome P450 superfamily.
In this study tested norendoxifen’s effects on the inhibition of important P450 enzymes as CYP19, CYP1A2, CYP2A6, CYP3A4, CYP3A5, and CYP2C19. In the presence of a range of concentrations of norendoxifen, the remaining enzyme activities of recombinant CYPs were determined by measuring the conversion rates from specific fluorometric substrates to their fluorescent metabolites. They demonstrated a high inhibitory potency of norendoxifen against CYP19 (aromatase) due to its lower half maximal inhibitory concentration (IC50= 131 nM). The IC50 values of E- and Z norendoxifen against recombinant CYP19 were 98 and 1053 nM, respectively suggesting that Norendoxifen has high isomer selectivity against CYP19. They also calculated the Ki value of Noredoxifen and E- and Z norendoxifen against aromatase obtaining a Ki value of 70, 48 and 445 nM, respectively. E-Norendoxifen had 9.3-fold-higher inhibitory ability than Z-norendoxifen against CYP19.
As a minor metabolite of tamoxifen, norendoxifen turns out to be the most potent aromatase inhibitor among the known metabolites that they have tested in previous studies. They demonstrate that norendoxifen is a potent and selective inhibitor of human aromatase with a Ki value in the nanomolar range (70 nM) as well as its IC50 (131 nM), while N-DMT and endoxifen act as AIs with IC50 values of 6.1 and 20.7 µM, respectively, via noncompetitive mechanisms.
The IC50 represents the concentration of a drug that is required for 50% inhibition and it is commonly used as a measure of antagonist drug potency in pharmacological research. Therefore, the lower the IC50 of the drug, the less will be need to achieve the desired effect, in other words the lower the IC50 the higher the potency of the drug. In the case of norendoxifen, it is almost 1000 fold time more potent that the other metabolites.
The findings obtained in this research are similar to another study, where norendoxifen inhibited recombinant aromatase via a competitive mechanism with a Ki of 35 nM and an IC50 of 90 nM, suggesting that this metabolite is much more potent inhibitor of aromatase than the other known inhibitory tamoxifen metabolites as endoxifen (IC50= 6 µM)and N-DMT (IC50= 15.9µM). According to Lu et al, the stepwise hydroxylation and demethylation of tamoxifen both resulted in progressive increases in inhibitory potency[7].
Due to the less amount of the norendoxifen needed to achieve a specific biological response and higher affinity (Ki) to the enzyme CYP19, we can suggest that norendoxifen has the most potent inhibitory ability against aromatase.
7. (10) Explain why step g of the P450 catalytic cycle is the most likely site for the reversible inhibition of lumarcoxib.
Lumiracoxib [2-(2-chloro-6-fluorophenyl)-amino-5-methylbenzeneacetic acid] was developed as a selective COX-2 inhibitor in the treatment of osteoarthritis, rheumatoid arthritis and acute pain. Lumiracoxib is metabolized predominantly by CYP2C9 with oxidation of the 5-methyl group and hydroxylation of the dihaloaromatic ring as the primary sites of biotransformation. Since lumiracoxib contains a 2-chloro-6-fluoroplenyl-amino group, the exposed 4-position on the aromatic ring was predicted to undergo metabolic activation by cytochrome P450 to a reactive quinoneimine intermediate, similar to the mechanism of cytochrome P450 mediated bioactivation of diclofenac. The bioactivation of lumiracoxib through quinoneimines may result in GSH depletion, covalent binding to proteins, oxidative stress, eventually leading to the observed hepatotoxicity[8].
The step g of the catalytic cycle is related to a hydrogen abstraction from the substrate by the electrophilic Compound I (Fe+4=0) that produces a carbon-centered radical perferric hydroxide complex. It could be also related to an electron abstraction from a heteroatom to form a heteroatom-centered radical cation perferryl intermediate. Metabolism of some substrates as lumiracoxib could result in the generation of reactive species that inactivates the enzyme through either covalent modification or tight binding.
A single electron transfer (SET) mechanism is involved an initial heteroatom oxidation to aminium cation radical, which could undergo the formation of highly reactive carbon-centered radical species and subsequently covalently bound to the amino residue within the enzyme active site.
Another mechanism that could explain the inhibition of CYP450 in the step g of the catalytic cycle is as follows: Lumiracoxib could be initially bound to Compound I through the N-H…O hydrogen-bonding interaction, the subsequent H-abstraction from the N-H bond results in an amino radical intermediate. Owing to the newly-formed hydrogen-bonding between O-H and N, dichotomous behaviors are encountered at the following step. The amino radical intermediates species could undergo the formation of C-centered radical species, which is essential for the enzyme inactivation[9].
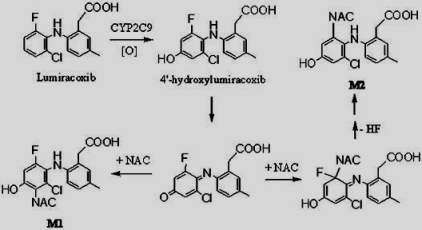
Figure 12. Proposed mechanism of Lumiracoxib by CYP2C9.[10]
8. (10) In Pamela’s seminar, (A Iwamura, KWatanabe, S Akai, TsNishinosono, et.al., Zomepirac Acyl Glucuronide Is Responsible for Zomepirac‐Induced Acute Kidney Injury in Mice. Drug Metabolism and Disposition July 2016, 44 (7) 888‐896), what is the significance of carboxylic acid drug safety in drug development.
Many compounds containing carboxylic acid functional groups are metabolized mostly in the liver through glucuronosyl transferase (UGT) enzyme family in addition to other metabolic pathways leading to the generation of metabolites. The UGT-mediated metabolism pathway is considered the most important for carboxylic acids and leads to the formation of acyl glucuronide metabolites, which are more polar than the parent compound with the hydrophilic glucuronic acid moiety. Carboxylic acid drugs are commonly found in nonsteroidal anti-inflammatory drugs (NSAIDs) as Zomepirac. However, the acyl glucuronides of carboxylic acid drugs tents to be highly reactive electrophiles, unstable under physiologic conditions and undergo hydrolysis or intramolecular rearrangements (migration of the drug moiety from the 1-O position to the 2, 3, 4 position on the glucuronide ring).
Carboxylic acids drugs are generally dissolved in a greater extent in the small intestine, where pH is favorable to ionized species. Other attractive properties of carboxylic acids as drug candidates include increased diversity of clearance mechanisms (introducing non-CYP mediated metabolism and biliary clearance, therefore, reducing CYP-mediated drug-to-drug interaction risk) and negatively charged moieties that can participate in salt bridges. The ability to form strong electrostatic interactions and hydrogen bonds is generally the major reason that carboxylic acids are selected as a functional group for drug-target interactions. However, charged compounds generally tend to have poor permeability and higher albumin binding, which can limit the volume of distribution and lower exposure to free drug[11].
Nevertheless, when glucuronidation takes place at carboxylate groups, the resulting bond is an ester-type linkage, which is much less stable and more reactive than the typical ether-linked glucuronides present in most glucuronidated phase 2 metabolites. Acyl glucuronides to be highly reactive can undergo hydrolysis, rapid intramolecular rearrangement and interact with normal cellular macromolecules (e.g., proteins, DNA, lipids) through covalent binding by transacylation or glycation. They may modify endogenous proteins and nucleophiles forming drug-protein adducts that can be recognized by the immune system as foreign, thereby eliciting an immune response. For instance, Zomepirac acyl glucuronide metabolite forms protein adducts with as Dipeptidyl peptidase IV, (cell membrane glycoprotein) that result in a toxic effect. Intrinsic reactivity is particularly important, since reactive metabolites and covalent binding have been associated with idiosyncratic toxic reactions, especially idiosyncratic hepatotoxicity. This has led some scientists in the pharmaceutical industry to argue that compounds with a carboxylic acid moiety should be avoided when developing new small molecule-based medicines[11][12]. Some nucleophilic molecules, such as GSH, generally represent an effective line of defense against endogenous and xenobiotic-derived electrophiles as acyl glucuronides.
Thereby is important to understand the various factors that can affect the safety of acyl glucuronide- drugs in drug development as stability of the acyl glucuronide, the chemical rate kinetics for the migration of the acyl group, and the concentration and stability/half-life of the antigenic protein.
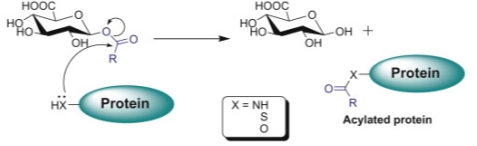
Figure 13. Proposed mechanisms for covalent binding of carboxylic acids to proteins via their acyl glucuronides. Nucleophilic displacement (acylation).[13]
9. (10). a. Can you provide an explanation why RO1 is not significantly metabolized by CYP450.
The RO1 compound is a p38 kinase inhibitor. The metabolite is produced by the addition of a hydroxyl group at the fourth carbon, this reaction is catalyzed by the aldehyde oxidase. The 4-hydroxylated metabolite is the major RO1 metabolite in humans and cleared through urinary excretion.The CYP450 enzymes seem not to be involved in the metabolism of this drug candidate, because this enzymes generally catalyze the oxidation of carbon atoms with a high electron density, and also in this case, the RO1 molecule presents aromatic deactivators as two molecules of fluorine in meta position. Halogens as fluorine are electron withdrawing groups leading to destabilize the intermediate cations. In contrast with aldehyde oxidase that catalyze the oxidation of electro-deficient double-bonded carbon atoms that are often present in nitrogen heterocycles as RO1 molecule.
b. Provide an explanation why RO1 is minimally metabolized by Phase 2 pathways
The clinical development of a candidate p38 kinase inhibitor was terminated because of its rapid clearance in human subjects. Aldehyde oxidase activity led to the rapid clearance of this novel p38 kinase inhibitor in human subjects, because this enzyme is in charge of the detoxification of different azaheterocycles. Thus, the drug is rapidly excreted by kidney and there is no need to conjugate the enzyme with any substance because is easily eliminated by urinary excretion. However, according to Zhang et al this drug could undergo a glucuronidation step.[14]
The chemical structure of RO1 appears to have a good affinity. There is no a handle, the enzyme eliminate faster o need to conjugation. A nucleophile is needed for conjugation )added by phse 1)
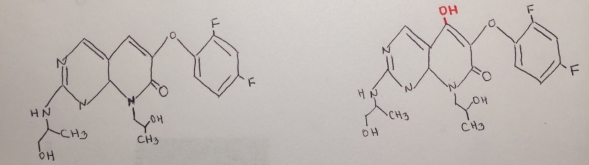
Figure 14. Reaction mechanism of the metabolite of RO1.
10. (10) Berberine is a natural alkaloid isolated from Coptis Chinensis. While this plant has been used in Ayurvedic and Chinese medicine for more than 2500 years, berberine is a widely used in non‐prescription dietary supplements for gastrointestinal infections and in treatment for diabetes and hypercholesterolemia. It has been suggested that drug-drug interactions between berberine‐containing products and CYP450 exist, but little is known about which CYPs mediate the metabolism of berberine in vivo.
a. Explain and show the mechanism of berberine CYP450 metabolism in liver microsomes
Berberine is an isoquinoline alkaloid that has multiple pharmacological activities, such as regulation of glucose and lipid, anti-bacterial, anti-inflammatory, anti-diarrheal, anti-tumor, anti-arrhythmia, immunosuppression and anti-cancer[15]. At least 3 major metabolites of Berberine have been identified in human blood after Phase I metabolism. Human CYP450 isoenzymes are responsible for BBR phase-I transformation remain to be identified. Berberine metabolites indicated that berberine phase I metabolites were M1 via oxidative demethylation, M2 via demethylenation, and M3 (jatrorrhizine), whereas its phase II metabolites were the corresponding glucuronide conjugates of M1, M2, and M3, respectively[16]. Figure 15 shows the proposed mechanism of berberine by CYP450:
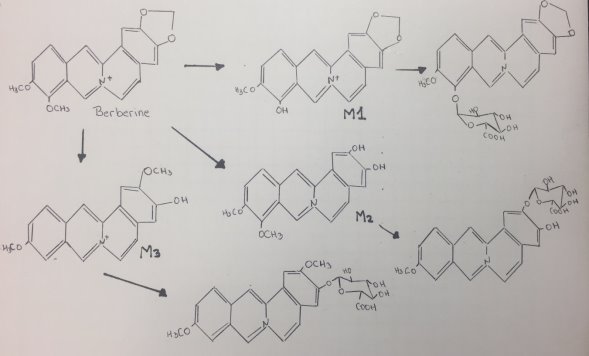
Figure 15. Proposed mechanism of Berberine.
b. Which human CYP450 isoforms are involved in the metabolism of berberine and are most likely involved with drug-drug interactions.
The metabolism of berberine was found to be catalyzed by several cytochromes P450 and UDP-glucuronosyltransferases in rat liver microsomes, whose major metabolic pathways were oxidative demethylenation and subsequent glucuronidation after intravenous administration. It has been demonstrated that CYP1A2, CYP2D6 and CYP3A4 are the major contributors for the transformation of berberine into its metabolites in human liver microsomes.[15]
However, the combination of berberine may cause drug–drug interactions (DDI) through induce or inhibit the metabolic enzyme and transporters in the present of disposition procedures of medicine such as metabolic enzymes profile and its gene transcription. It has been suggested that berberine can induce the activity of CYP enzyme, which is a result of the decrease of the bioavailability of drug such as verapamil, losartan, probenecid, cyclosporine A, cloxacillin, digoxin, pravastatin, midazolam and quinidine.
In a study performed by Cui et al investigated the metabolism of berberine and its effect on main metabolic enzymes in HepG2 cell. They used cocktail probe drugs as phenacetin (1A2), midazolam (3A4), tolbutamide (2C9) and chlorzoxazone (2E1). They also measured the mRNA expression and protein expression to evaluate the metabolism potency. Results showed that berberine could induce the activity of CYP1A2 and 3A4 in HepG2 cells, which is consistent with the increase of the enzyme gene transcription and protein expression. They found that this drug could significantly increase the metabolism of midazolam, phenacetin and tolbutamide with dose-dependence but no influence on metabolism of chlorzoxazone, the inducers of rifampicin and omeprazole could increase metabolic activity of CYP1A2 and 3A4 as well as inducer of BER. They concluded that berberine has the potential to induce CYP1A2 and 3A4 enzymes, which may lead to treatment failure such as midazolam, phenacetin and tolbutamide.[17]
In contrast, there is another study that conclude CYP2D6 was the primary recombinant human CYP producing these metabolites, followed by CYP1A2, 3A4, 2E1 and CYP2C19. The metabolism of berberine in human liver microsomes was decreased the most by a CYP2D inhibitor, and moderately by inhibitors of CYP1A and 3A. CYP2D6 plays a major role in berberine biotransformation, therefore, CYP2D6 pharmacogenetics and potential drug-drug interactions should be considered when berberine is used[18].
Cytochromes P450 are a family of enzymes which play a major role in the metabolism of drugs. However, just CYP1, 2 and 3 families are responsible for the biotransformation of most marketed drugs. One of the most important isoforms is CYP3A4 because is in charge to metabolize over 50% of drugs in clinical use. Some studies suggest that recombinant human CYP3A4 is responsible for formation of the major metabolite of berberine. The induction of CYP3A4 generate lower plasma levels than the concentration desired to achieve an effect, leading to therapeutic failure of several drugs. Therefore, there is a potential drug-drug interactions that should be considered when berberine is coadministered with drugs that are primarily metabolized by this isoform. Berberine is also metabolized by CYP1A2, which is responsible for the metabolism of several drugs. It has been demonstrated that berberine also induces CYP1A2 and affect the biotransformation of CYP1A2 substrates leading to numerous interactions with other drug. Finally, CYP2D6 is involved in the metabolism of berberine, this isoform metabolizes 20–25% of clinically used drugs and it is the most polymorphic isoform of all CYP450 inducing a potential drug interactions that should be considered when berberine or berberine-containing products are used.
Bonus Question (10 pts) after you complete questions 1‐10:
a. Explain the mechanism of oxidative metabolism of bergamottin at site A and C, but not site B.
Bergamottin, one of the major components in grapefruit juice, has been demonstrated to be responsible for the increased bioavailability of certain drugs in what has become known as the “grapefruit juice effect”. Bergamottin and its hydroxylated product 6′,7′-dihydroxy bergamottin (DHBG) are mechanism-based inactivators of CYP3A4. The ability of BG and DHBG to inactivate P450 3A4 is thought to be the major reason for the grapefruit juice-induced drug interactions that have been observed clinically. It have been reported that BG also inactivated P450 3A5 and 2B6, further underscoring the potential problems associated with this component and its effect on the metabolism of clinically important drugs[19].
The most easily oxidized region of the substrate near of the oxenoid compound and the most carbon center stable radical determine the regioselectivity of the enzyme. In the case of bergamottin, site C seems to be the one of the most likely sites of oxidation because this carbon is more substituted leading to a higher stability and reactivity. The two methyl groups are donating electrons to the radical and stabilizing the radical, also the charge will be spread out over more carbons. In contrast with B, that has a high degree of steric hindrance to the heme ferric-oxenoid, this fact could explain why this site is no favored to be oxidized. Site A is the furanocoumarin ring, this kind of ring tents to be oxidized in the furan double bond, leading to the formation of a furanoepoxide intermediate capable of combining with glutathione.
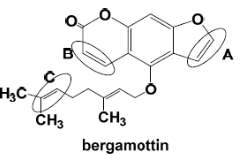
Figure 16. Chemical Structure of Bergamottin. D Williams
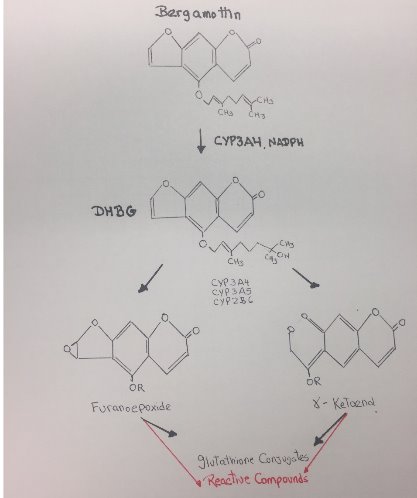
Figure 17. Reactive metabolites of Bergamottin.
b. Explain which site(s) leads to the formation of a reactive metabolite(s).
The metabolism of Bergamottin by P450 3A4/3A5 and 2B6 results in a loss in the enzymatic activity of these P450 through a combination of modification of the heme moiety and covalent modification of the apoprotein. Bioactivation of Bergamottin by these P450 generated reactive intermediates that inactivated these enzymes. Bergamottin has been shown to undergo hydroxylation of the phenyl ring, and oxidation of the furan ring with subsequent hydrolysis of the lactone ester. Subsequent studies established that one of the initial steps in the metabolism of furanocoumarins could involve oxidation of the furan double bond, leading to the formation of a furanoepoxide intermediate capable of combining either with glutathione to form the GS conjugate or with macromolecules.[20]
According to Kent et al, the most favorable binding site for Bergamottin/CYP2B6 interactions leads primarily to exposure of the terminal part of the geranyloxy chain and in particular the 5′-, 6′-, and 4′-carbons to the oxyferryl oxygen of compound I. These sites would be the most favorable sites of metabolism since they were the primary sites exposed of the active oxygen species[19].
The chemical structures of Bergamottin has a furanocoumarin ring structure, some studies demonstrated the formation of a monooxygenated metabolite of the furanocoumarin ring, the formation of a dihydrodiol furanocoumarin, and the oxidation of the double bond of the furan ring to generate a monooxygenated reactive intermediate that was responsible for inactivation and could be trapped by GSH[19].
In conclusion, the formation of a reactive intermediate that may bind to the heme or the apoprotein occurs on the furan ring of the drug via oxygen insertion at the furan double bond, leading to the formation of a reactive epoxide. Additional metabolism of Bergamottin by CYP450 involves hydroxylation on the geranyl-oxy moiety that also generates reactive compounds.
Bibliography:
[1] S.-E.W. Huskey, C. Zhu, M.M. Lin, R.R. Forseth, H. Gu, O. Simon, F.K. Eggimann, M. Kittelmann, A. Luneau, A. Vargas, Identification of three novel ring expansion metabolites of KAE609, a new spiroindolone agent for the treatment of malaria, in rats, dogs, and humans, Drug Metabolism and Disposition. 44 (2016) 653–664.
[2] P.R. Ortiz de Montellano, Hydrocarbon hydroxylation by cytochrome P450 enzymes, Chemical Reviews. 110 (2009) 932–948.
[3] L.L. Furge, F.P. Guengerich, Cytochrome P450 Enzymes in Drug Metabolism and Chemical Toxicology, Biochemestry and Molecular Education. 34 (2006) 66–74.
[4] F.P. Guengerich, Mechanisms of Cytochrome P450 Substrate Oxidation : MiniReview, 21 (2007).
[5] A.H. Meyer, A. Dybala-Defratyka, P.J. Alaimo, I. Geronimo, A.D. Sanchez, C.J. Cramer, M. Elsner, Cytochrome P450-catalyzed dealkylation of atrazine by Rhodococcus sp. strain NI86/21 involves hydrogen atom transfer rather than single electron transfer, Dalton Transactions. 43 (2014) 12175–12186.
[6] The Student Doctor Netwok, Noncompetitive vs allosteric inhibition: what’s the difference?, Forum. (n.d.). https://forums.studentdoctor.net/threads/noncompetitive-vs-allosteric-inhibition-whats-the-difference.942798/ (accessed April 24, 2017).
[7] W.J. Lu, C. Xu, Z. Pei, A.S. Mayhoub, M. Cushman, D.A. Flockhart, The tamoxifen metabolite norendoxifen is a potent and selective inhibitor of aromatase (CYP19) and a potential lead compound for novel therapeutic agents, Breast Cancer Research and Treatment. 133 (2012) 99–109. doi:10.1007/s10549-011-1699-4.
[8] A.P. Dickie, C.E. Wilson, K. Schreiter, R. Wehr, I.D. Wilson, R. Riley, Lumiracoxib metabolism in male C57bl/6J mice: characterisation of novel in vivo metabolites, (n.d.).
[9] X. Zhang, X.-X. Li, Y. Liu, Y. Wang, Suicide Inhibition of Cytochrome P450 Enzymes by Cyclopropylamines via a Ring-Opening Mechanism: Proton-Coupled Electron Transfer Makes a Difference, Frontiers in Chemistry. 5 (2017).
[10] Y. Li, J.G. Slatter, Z. Zhang, Y. Li, G.A. Doss, M.P. Braun, R.A. Stearns, D.C. Dean, T.A. Baillie, W. Tang, In vitro metabolic activation of lumiracoxib in rat and human liver preparations, Drug Metabolism and Disposition. 36 (2008) 469–473.
[11] T.R. Van Vleet, H. Liu, A. Lee, E.A.G. Blomme, Acyl Glucuronide Metabolites: Implications for Drug Safety Assessment, Toxicology Letters. (2017).
[12] M.P. Grillo, F. Hua, Identification of zomepirac-S-acyl-glutathione in vitro in incubations with rat hepatocytes and in vivo in rat bile, Drug Metabolism and Disposition. 31 (2003) 1429–1436.
[13] C. Li, L.Z. Benet, Mechanistic role of acyl glucuronides, Drug-Induced Liver Disease (Kaplowitz N and DeLeve L Eds) Pp. (2002) 151–181.
[14] X. Zhang, H.H. Liu, P. Weller, M. Zheng, W. Tao, J. Wang, G. Liao, M. Monshouwer, G. Peltz, In silico and in vitro pharmacogenetics: aldehyde oxidase rapidly metabolizes a p38 kinase inhibitor, The Pharmacogenomics Journal. 11 (2011) 15–24.
[15] Y. Li, G. Ren, Y.-X. Wang, W.-J. Kong, P. Yang, Y.-M. Wang, Y.-H. Li, H. Yi, Z.-R. Li, D.-Q. Song, Bioactivities of berberine metabolites after transformation through CYP450 isoenzymes, Journal of Translational Medicine. 9 (2011) 62.
[16] Y.-T. Liu, H.-P. Hao, H.-G. Xie, L. Lai, Q. Wang, C.-X. Liu, G.-J. Wang, Extensive intestinal first-pass elimination and predominant hepatic distribution of berberine explain its low plasma levels in rats, Drug Metabolism and Disposition. 38 (2010) 1779–1784.
[17] H.-M. Cui, Q.-Y. Zhang, J.-L. Wang, J.-L. Chen, Y.-L. Zhang, X.-L. Tong, In vitro studies of berberine metabolism and its effect of enzyme induction on HepG2 cells, Journal of Ethnopharmacology. 158 (2014) 388–396.
[18] Y. Guo, F. Li, X. Ma, X. Cheng, H. Zhou, C.D. Klaassen, CYP2D plays a major role in berberine metabolism in liver of mice and humans, Xenobiotica. 41 (2011) 996–1005.
[19] U.M. Kent, H. Lin, K.R. Noon, D.L. Harris, P.F. Hollenberg, Metabolism of bergamottin by cytochromes P450 2B6 and 3A5, Journal of Pharmacology and Experimental Therapeutics. 318 (2006) 992–1005.
[20] H. Lin, C. Kenaan, P.F. Hollenberg, Identification of the residue in human CYP3A4 that is covalently modified by bergamottin and the reactive intermediate that contributes to the grapefruit juice effect, Drug Metabolism and Disposition. (2012) dmd-112.
You have to be 100% sure of the quality of your product to give a money-back guarantee. This describes us perfectly. Make sure that this guarantee is totally transparent.
Read moreEach paper is composed from scratch, according to your instructions. It is then checked by our plagiarism-detection software. There is no gap where plagiarism could squeeze in.
Read moreThanks to our free revisions, there is no way for you to be unsatisfied. We will work on your paper until you are completely happy with the result.
Read moreYour email is safe, as we store it according to international data protection rules. Your bank details are secure, as we use only reliable payment systems.
Read moreBy sending us your money, you buy the service we provide. Check out our terms and conditions if you prefer business talks to be laid out in official language.
Read more