Abstract
Aims:
The aims of this project were to:
Methods:
Information was extracted from a number of different sources. All relevant information was assessed, analysed and presented using Microsoft Excel and Microsoft Word. A change control form was carried out as per SOP-QC-03-4.
µm- micrometres
A.niger- Aspergillus niger
AHU- Air Handling Unit
ASC- Adipose-derived Stem Cells
C.albicans- Candida albicans
CCF- Change Control Form
CCMI- Centre for Cell Manufacturing Ireland
Cfu- colony forming units
CLI- Critical Limb Ischemia
DNA- deoxyribonucleic acid
DQ- Design Qualification
E.coli- Escherichia coli
E.hirae- Enterococcus hirae
EM- Environmental Monitoring
EU- European Union
GMP- Good Manufacturing Practice
GNC- Gram Negative Cocci
GNR- Gram Negative Rod
GPC- Gram Positive Cocci
GPR- Gram Positive Rod
HEPA- High Efficiency Particulate Air
HMSCs- Human Mesenchymal Stem Cells
HPRA- Health Products Regulatory Authority
IPA- Isopropyl Alcohol
IQ- Installation Qualification
ISO- International Standards Organisation
kGy- kilogray
m3 – meters cubed
MF- Media Fill
NCBES- National Centre for Biomedical Engineering Science
OQ- Operation Qualification
PQ- Production Qualification
Ps.aeruginosa- Pseudamonas Aeruginosa
R&D- Research and Development
S.aureus- Staphylococcus aureus
SOP- Standard Operating Procedure
TSA- Trypticase soy agar
URS- User Requirement Specification
Table of Contents
1.1 Centre for Cell Manufacturing Ireland
1.4 EudraLex Volume 4, Annex 1: Manufacture of Sterile Medicinal Products 2008
1.5 IS0 14644:2015: The Standard for Cleanrooms and Associated Controlled Environments
1.7 Cleanroom Qualification and Validation
1.9 Cleanroom Contamination and Contamination Control
1.10 Gowning and Cleanroom Personnel
Chapter 2: Methods and Materials
2.1 Assessing 2016 Cleaning Records:
2.2 Assessing Environmental Monitoring Trends for Batches carried out in 2016:
2.4 An investigation into the Gram Reaction of Organisms Found:
2.5 An investigation into the Microbial Identification of Organisms Found:
2.6 An Investigation into the Disinfectants Used in the CCMI:
3.2 EM Results from Batch CLI-16-001
3.3 EM Results from Batch CLI-16-002
3.4 EM Results from Batch ASC-16-001
3.5 EM Results from Batches MF-ASC 16-001, MF-ASC 16-002 & MF-ASC 16-003
3.7 Gram Stain Results from all Microorganisms Detected growing on TSA plates in EM.
Appendix A- Example of an EM Recording and Reporting Form
Appendix B- Example of a Microbial Identification Form
Figure 1: Example of the differentiation of a stem cell into a number of
Different independently functioning cell…………………………………………….……3
Figure2:Percentage of each Gram Reaction………………………………….……..…19
Figure 3: Percentage of each Microorganism found in Grade A and B Areas…………25
Figure 4: Percentage of Time each Biocide was used for All Cleaning Records………26
Table 1: Maximum permitted number of airborne particle per cubic meter for classification of each grade cleanroom as per EudraLex Volume 4, Annex 1……………………………….5
Table 2: Maximum Concentration Limits (Particles/m3) of each IS0 14644-1:2015 Classification Number. (ISO Class 1-ISO Class 9)……………………………………………6
Table 3: ≥ 0.3 µm Particles Shed during Various Activities………………………….…..…10
Table 4: Cleaning Records for 2016 Showing: Date, Type of Cleaning and Biocide
Used……………………………………………………………………………………………18
Table 5:EM Results from Batch CLI-16-001…………………………………………………20
Table 6: EM Results from Batch CLI-16-002……………………………………………….21
Table 7: EM Results from Batch ASC-16-001………………………………………………22
Table 8:EM Results from Batches MF-ASC 16-001 & MF-ASC 16-002 & MF-ASC 16-003……………………………………………………………………………………………23
Table 9:Microbial Identification and Gram Reaction of Organisms Found growing on TSA Plates in Batches; CLI 16-001, CLI 16-002, ASC 16-001, MF-ASC 16- 001, MF-ASC 16-002 & MF-ASC 16-003…………………………………………….……………………………..24
Table 10: Number of each Gram Reaction……………………………………………………25
Table 11: Microbial Identification, Number and Gram Reaction of Organisms found in Grade A and B Areas………………………………………………………………………….26
Table 12: Description, Instructions for Use and Mode of Action for Premier-Klercide 70/30 IPA……………………………………………………………………………………………27
Table 13: IPA efficacy testing summary……………………………………………….…….28
Table 14: Description, Instructions for Use and Mode of Action for Premier Klercide-CR Biocide A……………………………………………………………………………………..29
Table 15: Biocide A efficacy testing summary………………………………………………30
Table 16:Description, Instructions for Use and Mode of Action for Premier Klercide-CR
Biocide B……………………………………………………………………………………..31
Table 17: Biocide B efficacy testing summary………………………………………………33
Table 18: Description, Instructions for Use and Mode of Action for Premier Klercide-CR Biocide C……………………………………………………………………………………..34
Table 19: Biocide C efficacy testing summary………………………………………………35
The Centre for Cell Manufacturing Ireland (CCMI) is a research facility housed in the National Centre for Biomedical Engineering Sciences (NCBES), both are part of the National University of Ireland, Galway. It is the first and only centre for stem cell manufacturing in Ireland for use in human clinical trials. The centre received authorisation by the Health Products and Regulatory Authority (HPRA) in 2013. The CCMI are manufacturing Human Mesenchymal Stem Cells (hMSCs) which are derived from human bone marrow and these are used in a number of different clinical trials for the treatments of conditions such as heart disease, diabetes, and arthritis. The facility itself contains two production suites, one of which is not in use. Each suite has 3 processing rooms, each certified to an EU GMP grade A, B, C or D, therefore allowing for the aseptic production of a number of different batches of these advanced therapeutics. (Finnerty 2017)
Stem cells are self-renewing and are known as progenitor cells, meaning they have the ability to differentiate into any cell type by asymmetric mitosis which allows the synthesis of two intrinsically different daughter cells. They can divide rapidly and continuously. The stem cells are a high plasticity in that they have a complex ability to cross lineage barriers and adopt the expression profile and functional phenotypes of the cells that are typical of other tissues. Phenotype refers to the physical properties of a cell. This “plasticity” can be further explained by incorporating the terms transdifferentiation and fusion. Transdifferentiation is the attainment of the identity of a different phenotype through the expression of the gene pattern of other tissues or through the achievement of a more primitive state and the successive differentiation to another cell types. Fusion involves the union of a stem cell with a cell of another tissue; a cell can express a gene and acquire a phenotypic element of another parenchyma. A parenchyma is the functional tissue of an organ as distinguished from the connective and supporting tissue. (Lodi et al 2011)
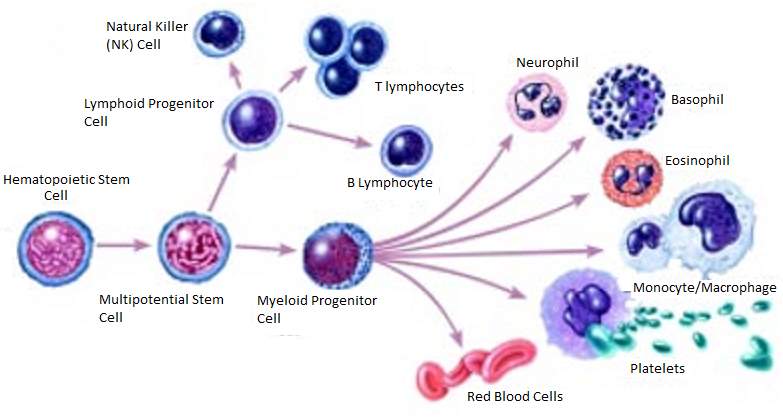
Figure 1: Example of the Differentiation of a Stem Cell into a number of Different Independently Functioning Cells. (National Institutes of Health 2016)
The term biopharmaceuticals refers to the class of medicinal products that are produced in living cells, whereas pharmaceuticals are produced chemically. Biologic biopharmaceuticals are generally larger in size in comparison with chemically synthesised pharmaceuticals; they can be produced in a variety of different forms e.g. therapeutic vaccines, nucleic acid preparations, and recombinant therapeutic proteins. The production of these biopharmaceuticals began in the 1970s with the emergence of genetic engineering and cell culture as new technologies. Genetic engineering allowed researchers to create specific recombinant DNA molecules encoding specific proteins. Cell culture was then used to introduce these genetically engineered cells into bacterial or animal cells to synthesis the protein of interest. Cell culture permits the development of highly viable, high-density cell cultures in large volumes.
Biopharmaceuticals are a growing part of the R&D pipeline across the pharmaceutical industry with more and more new biologics candidates being discovered. Biologics that have gained regulatory approval over the past 30 years include molecules that can treat a variety of diseases, allowing the root cause of these diseases to be treated, rather than just easing the symptoms.
As biologics are proteins, they cannot be sterilised by heat as they could be denatured and lose their function, for this reason, they are produced aseptically within cleanroom under the most stringently controlled conditions in order to prevent the product from becoming contaminated. The most common manufacturing processes used are perfusion or fed-batch. Perfusion involves the continuous addition nutrients and removal of waste (medium containing metabolic waste) and the desired product without stopping the process. Fed-batch allows for the addition of nutrients but requires the process to be stopped to remove waste and the desired product.
In recent times the manufacturing process of biopharmaceutical has progressed to one of high complicity with state-of-the art operations. Modern facilities boost multiple departments that function as one to produce, test and assure quality of these biopharmaceuticals before they can be released for further processing. The product must be presented as a sterile substance; it may be lyophilised if a solid or stored in vials or syringes if a liquid. Good Manufacturing Practice (GMP) is followed throughout the entire process to ensure that the highest quality and highest quality product is released to the public- more than often an immunocompromised individual. (Hughes and Hann 2007)
The EudraLex is the documentation containing the rules governing medicinal products within the European Union. In contains details on the manufacture of sterile products, which is subject to “special requirements in order to minimize risks of microbiological contamination, and of particulate and pyrogen contamination.” The production of such products relies on the skill, training and attitudes of the personnel involved. Quality Assurance is of the utmost importance. Therefore this type of manufacturing must stringently follow carefully established and validated methods of preparation. Sole reliance for sterility or other quality aspects must not be placed on any terminal process or finished product test. The document contains 127 subsections detailing the manufacture of sterile medicinal products under the following heading; General, Cleanroom and clean air device monitoring, Isolator technology, Blow/fill/seal technology, Terminally sterilised products, Aseptic preparation, Personnel, Premises, Equipment, Sanitation, Processing, Sterilisation, Sterilisation by heat, Moist heat, Dry heat, Sterilisation by radiation, Sterilisation with ethylene oxide, Filtration of medicinal products which cannot be sterilised in their final container, Finishing of sterile products, Quality control. Cleanrooms are classified in accordance with this document. This is done so by assessing the airborne particle concentration within an area and comparing it to this following table:
Table 1: Maximum permitted number of Airborne Particle per cubic meter for Classification of Each Grade Cleanroom as per EudraLex Volume 4, Annex 1. (EudraLex 2008)
| Grade | Maximum permitted number of particles per m3 equal to or greater than the tabulated size | |||
| At rest | In operation | |||
| 0.5 µm | 5.0µm | 0.5 µm | 5.0µm | |
| A | 3,520 | 20 | 3520 | 20 |
| B | 35,200 | 29 | 352,000 | 2,900 |
| C | 353,000 | 2,900 | 3,520,000 | 29,000 |
| D | 3,520,000 | 29,000 | Not defined | Not defined |
ISO 14644:2015 is the latest edition of ISO 14644 which sets. This standard consists of 13 different parts including: Classification of air cleanliness, Classification of air cleanliness by particle concentration, Specifications for testing and monitoring to prove continued compliance with ISO 14644-1, Monitoring to provide evidence of cleanroom performance related to air cleanliness by particle concentration, Test Methods, Design, construction, and start-up, Operations, Vocabulary, Separative devices (clean air hoods, gloveboxes, isolators and minienvironments), Classification of airborne molecular contamination, Classification of surface particle cleanliness, Classification of Surface Cleanliness by Chemical Concentration and Classification of Air Cleanliness by Nanoscale Particle Concentration.
Each of the subsections stated above details the appropriate guidelines to be followed in order to comply with Class 1-Class 9 cleanroom requirements. The following demonstrates the current acceptable concentration of particles permitted within each class of cleanroom according to ISO 14644. It was be seen that, in comparison with Eudralex Volume 4, Annex 1: Manufacture of Sterile Medicinal Products 2008- described in the section 1.3, that with regards to the 0.5 µm particle concentration, Grade A, B C and D have the same conditions as Class 5,6,7 and 8 (in the ISO 14644 classification).
Table 2: Maximum Concentration Limits (Particles/m3) of each IS0 14644-1:2015 Classification Number. (ISO Class 1-ISO Class 9)
| ISO 14644-1:2015 Classification Number (N) | Maximum Concentration Limits (Particles/m3) | |||||
| 0.1 µm | 0.2 µm | 0.3 µm | 0.5 µm | 1.0 µm | 5.0 µm | |
| ISO Class 1 | 1 | 10 | ||||
| ISO Class 2 | 100 | 24 | 10 | |||
| ISO Class 3 | 1,000 | 237 | 102 | 35 | ||
| ISO Class 4 | 10,000 | 2,370 | 1,020 | 352 | 83 | |
| ISO Class 5 | 100,000 | 23,700 | 10,200 | 3,520 | 832 | |
| ISO Class 6 | 1,000,000 | 237,000 | 102,000 | 35,200 | 8,320 | 298 |
| ISO Class 7 | 352,000 | 83,200 | 2,930 | |||
| ISO Class 8 | 3,520,000 | 832,000 | 29,300 | |||
| ISO Class 9 | 35,200,000 | 8,320,000 | 293,000 | |||
Cleanroom definition: a confined area in which the humidity, temperature, particulate matter, and contamination are precisely controlled within specified parameters. (Chemicool 2017) Cleanrooms are facilities used to create a controlled environment in manufacturing and scientific research where small particle contamination can have an adverse effect on a product or manufacturing process for example; the biopharmaceutical, pharmaceutical, aerospace, and medical device industry. Cleanroom are designed in such a way as to ensure low levels of particles (pollutants) e.g. airborne microbes, aerosol particles and various vapours. In order to categorise a cleanroom to a certain glade or class a controlled level of contamination at each relevant particle size per cubic meter must a retained. As stated in section 1.3 and 1.4.
The key component used to control particulate matter is High Efficiency Particulate Air (HEPA) filters. These are used to filter the air entering the facility, the can remove particles ≥ 0.3 µm. Air Handling Units (AHU) containing these HEPA filters also control the temperature of the air circulating the facility and the number of air changes through the control of air ducts. The clean air is circulated through the facility using a unidirectional air flow system, the air flows in a horizontal or vertical pattern over and around any objects including personnel within the cleanroom. A high number of air changes- up to 60/hr. is needed to ensure high levels of clean air is being used. Airlocks and differential pressure cascades are used to decrease the number of particles entering the cleanroom areas. (Clean Air Technology, Inc. 2017)
There is a formal and documented qualification and validation process for the design, building and qualification of a new cleanroom. This validation and qualification is undertaken to prove that all components of the cleanroom consistently do as they are supposed to. Under GMP guidelines each facility must prove control of the critical aspects of certain operations which requires testing and documentation. This is carried out to ensure that the equipment can perform consistently and that it meets the process parameters.
Validation is generally carried out in accordance with a validation master plan; this states how the validation must be carried out in a clear and easily understood manner. In the validation process the following aspects are taken into consideration:
• User requirement specification (URS), this states the user’s objective for the product.
• Design qualification (DQ), where the design of the product is compared with the appropriate standards and requirements.
• Installation qualification (IQ), this involves the testing of the product to verify that it is installed correctly.
• Operational qualification (OQ), which is testing to verify that the equipment operates as stated.
• Performance qualification (PQ), this is verification that the product can be consistently produced to specification.
Cleanrooms are planned and manufactured using strict protocol and methods. When designing a cleanroom the one aspect that everything related back to is contamination control. The initial concepts to be established include; the area of the cleanroom, the grade of cleanroom required, the number of air changes required, the pressure cascade required, whether isolator technology will be included, how to include a maximum number of HEPA filters, minimising air flow resistance, the properties of the ductwork, adapting air flow when in different states, e.g. at rest, in occupation and finally defining the electrical systems needed.
Qualification and validation are undertaken to prove that each component of the cleanroom is working as it was specified to on a consistent basis. The performance of the cleanroom refers to all the interactions within the cleanroom, including; airflow, sources of contamination, sources of heat, positions of vents, exhausts and other objects occupying the space. If changes are made to any of these components, there will possibly be a knock on effect on the operation of the cleanroom and could undermine other aspects of the cleanroom design.
In order to reduce contamination, the cleanroom is constructed in such a way that they are easy to clean and disinfect. They are constructed with a smooth and cleanable finish, have a finishing coat that is impervious to detergents and disinfectants, no uncleanable recesses, smooth curves and integrated services. (Sandle 2014)
Cleanroom contamination is mostly focuses of sub-micron airborne particles, they are kept to a minimum through a process of control, and they must be continually removed from the air. Contamination causes materials or surfaces to be soiled with contaminating substances. The main sources of contamination in a cleanroom are personnel, facility, tool generated, and raw material. These sources need to be controlled and eliminated, where possible as they can be the source for “killing a circuit”. Prevention of these contaminants from entering the cleanroom is the main object in contamination control. Temperature, humidity, air flow direction, air changes and specialised filtration are all rigorously controlled. Commitment from any personnel entering the cleanroom is required. Cleanroom cleaning is also an essential part in the maintenance of the specific cleanroom required for a process. (McFadden 2017)
Gowning is used to control the spread of non-viable particulate and viable micro-organisms within a cleanroom environment. These viable and non-viable particulates must be controlled to reduce the risk of introducing contamination to a process. Cleanroom personnel are the main source of contamination within the cleanroom, they are responsible for 80-90% of contamination present, therefore it is essential to incorporate controlled change procedures and the use of low-particulate cleanroom garments to reduce this contamination. It is essential the correct size garments are available for staff at all time to prevent poorly fitting garments from allowing the spread of infection.
The type of gowning to be implemented by a company depends on: regulatory guidelines, the risk of microbial/particulate contamination, the classification of the cleanroom, the operations being carried out and the risk of the process to the personnel. Standard Operating Procedures (SOPs) must be established, understood and followed.
All personnel must undergo gowning qualifications before they can enter a cleanroom area to avoid the risk of contamination entering the cleanroom as a result of poor gowning. The gowning qualification involves the examination of an employee’s aseptic technique when gowning up, contact plates are used at the end of the process to assess an employee’s ability to aseptically carry out the process. (Cleanroom Technology 2015)
When personnel are inside the cleanroom, their physical actions and psychological attributes can cause negative effects on the environment. Psychological concerns for example room temperature, humidity, odours and work attitude all have an effect on how the personnel work within the cleanroom and also on the concentration of particles in the cleanroom. The personnel’s physical presence alone in the cleanroom adds to the particle count as seen in the table on the next page.
Table 3: ≥ 0.3 µm Particles shed while Standing/Sitting, Walking and Horseplay.
| Activity | Particles/Minute (≥ 0.3 µm) |
| Standing/Sitting | 100,000 |
| Walking | 5,000,000-10,000,000 |
| Horseplay | 100,000,000 |
Cleaning is an essential process in contamination control. Cleaning procedures must be established and followed. Cleaning may be carried out by the facility personnel or by a contractor company. It is essential that whoever is responsible from the cleaning is aware of and understands the current procedures. The following are important aspects of a cleaning procedure that need to be answered before a cleaning programme can be established; how is the cleaning assessed, what cleaning materials should be used, when can/should the cleanroom be cleaned?
Although filtration, design and training of personnel are all established to prevent contamination within the cleanroom it is still imperative to ensure that a good cleaning programme has been established for a facilities cleanroom. The cleaning procedure used from company to company will differ depending on a number of variables. A written SOP should be established and understood by all personnel partaking in the cleaning programme. The frequency of cleaning should also be established and followed stringently.
In order to establish a cleaning programme the following should be taken into account: Environmental Monitoring (EM), which cleaning chemicals/disinfectants should be used based on EM results, evaluating the appropriate cleaning materials for user and cleanroom class, carry out verification/ efficacy tests, train personnel and partake in continued compliancy checks. (DiDonna, 2012)
Rotational cleaning is now a common approach used, it involves the use of at least 2 cleaning products- one being a disinfectant and the other being sporicidal, as stated in the EudraLex Annex 1 – Manufacture of sterile medicinal products – Volume 4 EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary use, “The sanitation of clean areas is particularly important. They should be cleaned thoroughly in accordance with a written programme. Where disinfectants are used, more than one type should be employed. Monitoring should be undertaken regularly in order to detect the development of resistant strains”. (EudraLex 2008) This helps to prevent the growth of bacterial spores and to reduce the bioburden level in the cleanroom area. (Polarine and Rogers 2014)
The fundamental purpose of rotation is to prevent selection for resistant organisms; not to prevent organisms from becoming resistant to disinfectants, an idea which is widely believed. A good rotational cleaning programme should include products which control a wide variety of organisms, complementing each other, giving a synergistic effect. (Sartain 2005)
As there is little to no scientific evident to support the idea of disinfectant resistance, it is a regulatory expectation for companies to adhere to the concept of rotational cleaning. The frequency of rotation is based upon EM trend results. (Cleanroom Technology 2010)
Computer Software:
CCMI Documentation:
External Documentation:
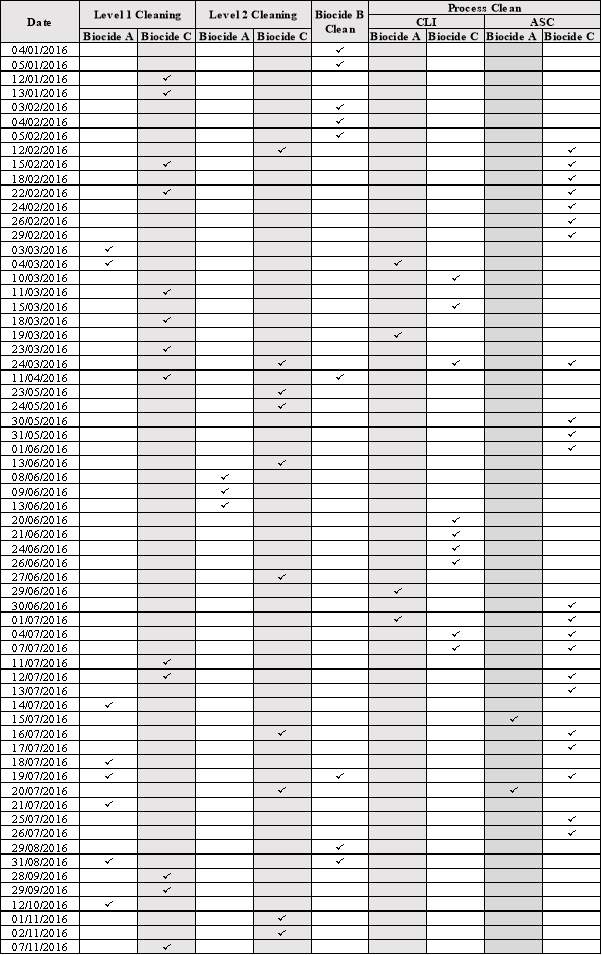
Table 4: Cleaning Records for 2016 Showing: Date, Type of Cleaning and Biocide Used.
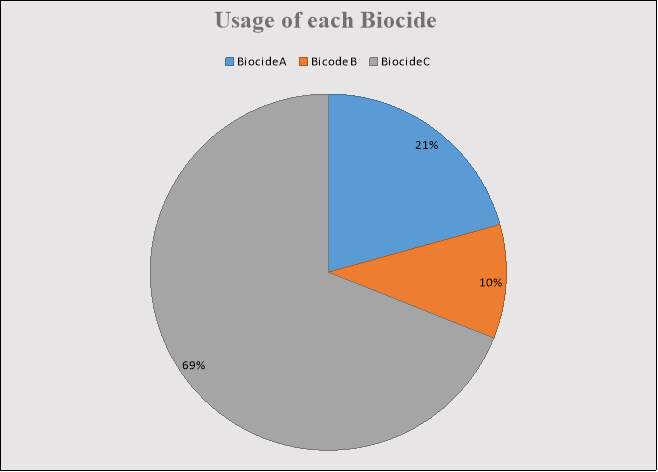
Figure 2: Percentage of Time each Biocide was used.

Table 5:EM Results from Batch CLI-16-001

Table 6: EM Results from Batch CLI-16- 002

 Reaction of Organisms Found in Batches; CLI-16-001,CLI-16-002, ASC-16-001, MF-ASC 16- 001,MF-ASC-16-002 & MF-ASC-16-003
Reaction of Organisms Found in Batches; CLI-16-001,CLI-16-002, ASC-16-001, MF-ASC 16- 001,MF-ASC-16-002 & MF-ASC-16-003
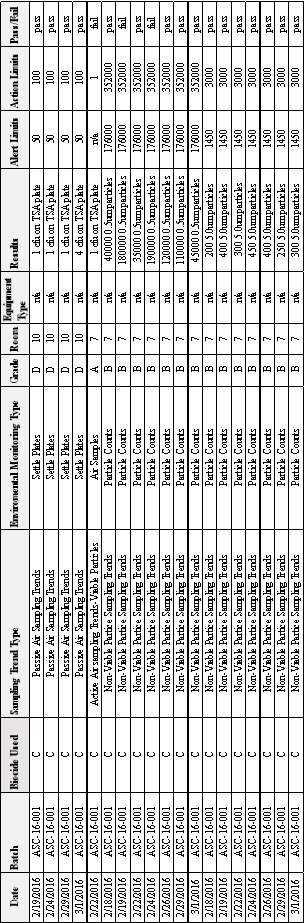
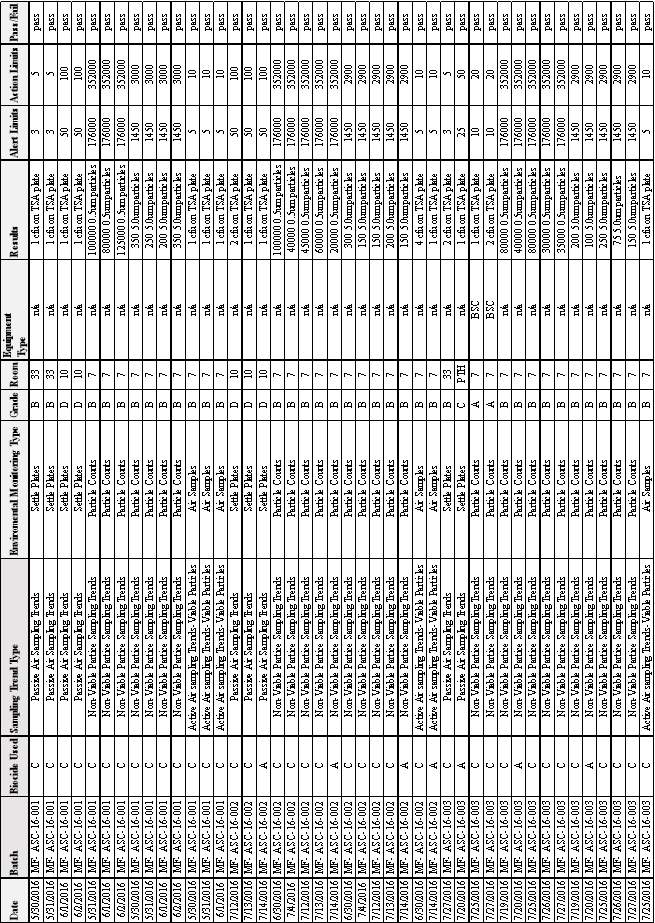
Table 8:EM Results from Batches MF-ASC 16-001 & MF-ASC 16-002 & MF-ASC 16-003
Table 7: EM Results from Batch ASC-16- 001
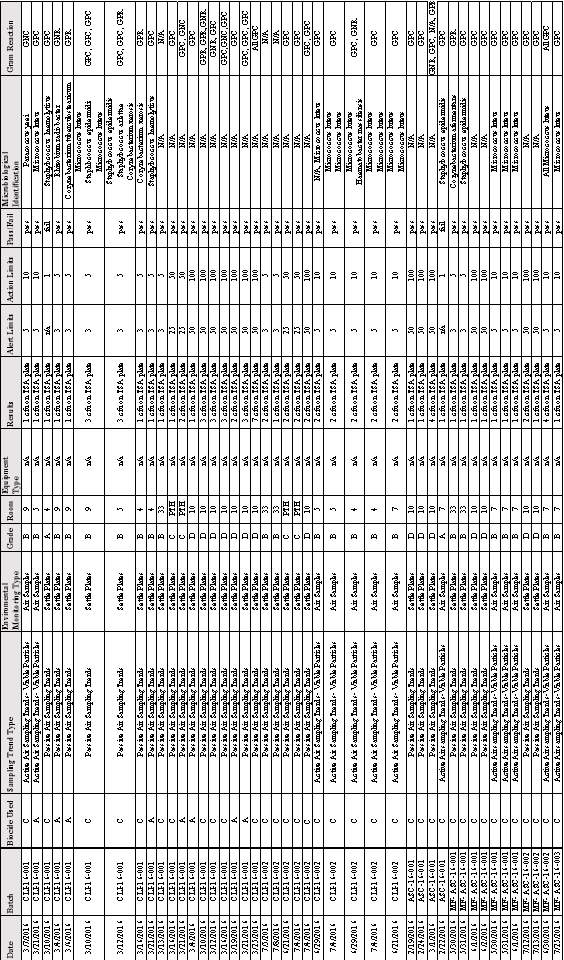
3.6 Microbial Identification and Gram Reaction of Microorganisms Found Growing on TSA Plates All Batches
Table 9:Microbial Identification and Gram Reaction of Organisms Found growing on TSA Plates in Batches; CLI 16-001,CLI 16-002, ASC 16-001, MF-ASC 16- 001,MF-ASC 16-002 & MF-ASC 16-003
 3.7 Gram Stain Results from all Microorganisms Detected growing on TSA plates in EM.
3.7 Gram Stain Results from all Microorganisms Detected growing on TSA plates in EM.Table 10: Number of each Gram Reaction.
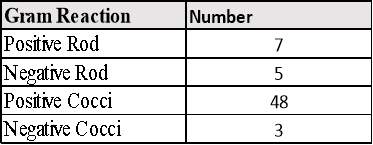
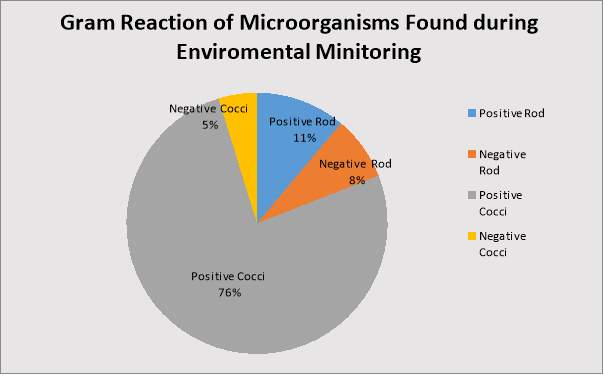
Figure 4: Percentage of each Gram Reaction.
Table 11: Microbial Identification, Number and Gram Reaction of Organisms found in Grade A and B Areas.
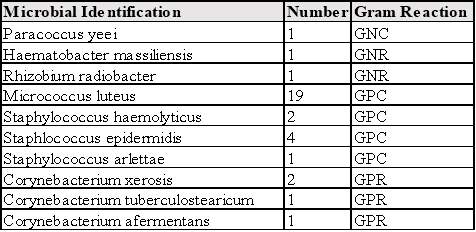
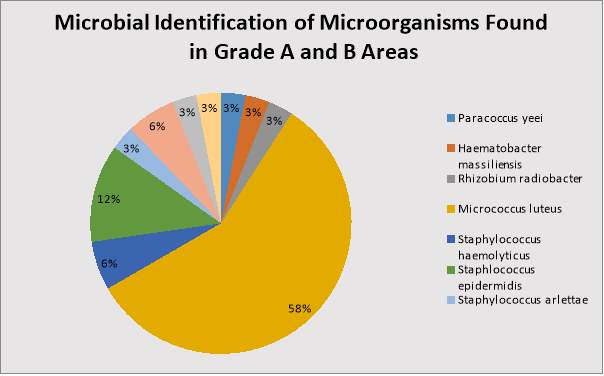
Figure 5: Percentage of each Microorganism found in Grade A and B Areas.
Table 12: Description, Instructions for Use and Mode of Action for Premier-Klercide 70/30 IPA.
| Description | Instructions for Use | Mode of Action | |
| IPA |
|
|
Microbiocidal against bacteria e.g. mycobacteria, fungi and lipids containing viruses. Has the ability to denature proteins by attacking their cell membrane.
Alcohols change the configuration of proteins and prevent them from performing their function.
When lipids in the cell membrane come into contact with 50-80% alcohol solutions the surface tension of the molecule is lowered which allows extra cellular water present in the surrounding environment to pass via osmosis through the cell membrane, causing bacterial lysis. |
IPA Efficacy Testing
The following European Standards were used to test the efficacy of Premium Klercide- 70/30 Sterile IPA to show good broad-spectrum biocidal activity. The results have been summaries in this table.
Chemical Disinfectants and Antiseptics- Quantitative suspension test for the evaluation of bactericidal activity of chemical disinfectants and antiseptics used in food, industrial, domestic and institutional areas.
Chemical Disinfectants and Antiseptics- Quantitative suspension test for the evaluation of fungicidal activity of chemical disinfectants and antiseptics used in food, industrial, domestic and institutional areas.
Chemical Disinfectants and Antiseptics- Quantitative non-porous surface test for the evaluation of bactericidal and/or fungicidal activity of chemical disinfectants used in food, industrial, domestic and institutional areas.
Table 13: IPA efficacy testing summary.
| Organism | Pass Criteria (Log Reduction) | Contact time (minutes ±10 s.) | Temperature (±1 C°) | Result (Log Reduction) | Pass/Fail | |
| Clean | Dirty | |||||
| BS EN 1276:1997 | ||||||
| Ps.aeruginosa | 5 | 5 | 20 | >6.9 | >6.9 | Pass |
| E.coli | 5 | 5 | 20 | >7.0 | >7.0 | Pass |
| E.hirae | 5 | 5 | 20 | >6.7 | >6.7 | Pass |
| S.aureus | 5 | 5 | 20 | >6.7 | >6.7 | Pass |
| BS EN 1650:1998 | ||||||
| A.niger | 4 | 15 | 20 | 3.9 | >4.7 | Pass |
| C.albicans | 4 | 15 | 20 | >5.7 | >5.7 | Pass |
| BS EN 13697:2001 | ||||||
| Ps. Aeruginosa | 4 | 5 | 18-25 | >4.1 | N/A | Pass |
| E.coli | 4 | 5 | 18-25 | >4.5 | N/A | Pass |
| E.hirae | 4 | 5 | 18-25 | >5.6 | N/A | Pass |
| S.aureus | 4 | 5 | 18-25 | >6 | N/A | Pass |
| C.albicans | 3 | 5 | 18-25 | >4.1 | N/A | Pass |
| A.niger | 3 | 15 | 18-25 | >4.7 | N/A | Pass |
| M.luteus | 5 | 5 | 18-25 | >5.7 | N/A | Pass |
| St.capitis | 5 | 5 | 18-25 | >5.8 | N/A | Pass |
| S.hominis | 5 | 5 | 18-25 | >5.4 | N/A | Pass |
| Pen.compactum | 4 | 15 | 18-25 | >4.9 | N/A | Pass |
Table 14: Description, Instructions for Use and Mode of Action for Premier Klercide-CR Biocide A.
| Biocide | Description | Instructions for Use | Mode of Action |
| A |
|
Formulated to be used as broad spectrum disinfectant at a 1:3 dilution.
|
Some penetrate the cell wall, entering the cell cytoplasm, destroying protein groups essential for life. Surfactants damage the cell by reducing its permeability, disturbing the normal flow of nutrients into the cell and the discharge of its waste. Cationic surfactants, e.g. quaternary ammonium compounds, adsorb to the cell membrane, reacting chemically with the negatively charged ions associated with the walls. Anionic surfactants reduce cell permeability and dissolve the entire membrane.
Alkyl benzyl ammonium chloride is a cationic detergent, which is effective against vegetative bacteria and some fungi, but not against spores and mycobacteria. They can be inactivated by proteins, a variety of natural and plastic materials, anion detergents and soaps. They have the advantage of being stable and non-corrosive. The target of this biocide is the bacterial cell envelope. The quaternary ammonium compound is absorbed onto the bacterial cell surface. It diffuses through the cell wall and binds to the cytoplasmic membrane, disrupts it, which causes the cytoplasmic constituents to be released. This results in the death of the cell. |
Biocide A Efficacy Testing
The following European Standards were used to test the efficacy of Premium Klercide-CR Biocide A to show good broad-spectrum biocidal activity. The results have been summaries in this table.
Chemical Disinfectants and Antiseptics- Quantitative suspension test for the evaluation of bactericidal activity of chemical disinfectants and antiseptics used in food, industrial, domestic and institutional areas.
Chemical Disinfectants and Antiseptics- Quantitative suspension test for the evaluation of fungicidal activity of chemical disinfectants and antiseptics used in food, industrial, domestic and institutional areas.
Chemical Disinfectants and Antiseptics- Quantitative non-porous surface test for the evaluation of bactericidal and/or fungicidal activity of chemical disinfectants used in food, industrial, domestic and institutional areas
Table 15: Biocide A efficacy testing summary.
| Organism | Pass Criteria |
You have to be 100% sure of the quality of your product to give a money-back guarantee. This describes us perfectly. Make sure that this guarantee is totally transparent.
Read moreEach paper is composed from scratch, according to your instructions. It is then checked by our plagiarism-detection software. There is no gap where plagiarism could squeeze in.
Read moreThanks to our free revisions, there is no way for you to be unsatisfied. We will work on your paper until you are completely happy with the result.
Read moreYour email is safe, as we store it according to international data protection rules. Your bank details are secure, as we use only reliable payment systems.
Read moreBy sending us your money, you buy the service we provide. Check out our terms and conditions if you prefer business talks to be laid out in official language.
Read more