Every year, thousands of chemical and biological entities are developed in the hope of making it to the shelf. The drug development process is the progression by which drugs will either be approved or not approved for use, and spans from discovery and preclinical testing to Phase IV of Clinical Trials (post marketing surveillance). The underpinning aim is to ensure the safety and efficacy of a drug before it can be approved for use. Given that only one in every 5,000 to 10,000 compounds that enter preclinical testing are approved for marketing, and that an average of 15 years is required to take a drug through the complete process, the drug development process is a long and costly one.
The two main regulatory bodies in the world are the FDA (USA) and the EMA (EU) and while the drug approval process varies from the EU to the USA, the basic regulation process remains the same.
Figure 1 Basic drug development regulation
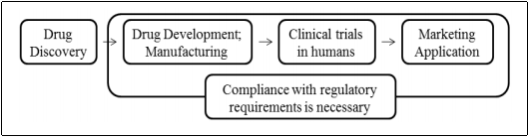
(Kashyap et al., 2013)
In brief, the process of drug approval in the US first involves the submission of an Investigational New Drug application by the Sponsor. This allows the Clinical Trials to commence once the data from the Preclinical trials shows that the drug is safe. Once the drug is shown to be safe and effective (Clinical Trials data), a New Drug Application is filed which is essentially an application to manufacture and sell the drug (Rick, 2009).
The application process for the EU, similar to the US, requires two steps. The first step is a clinical trial application to an individual member state, and then a marketing approval. The marketing approval in the EU can take one of three approaches
As mentioned before, the purpose of the drug development process is to ensure safe and effective drugs are available to the patient. However, both the US and EU processes are very lengthy procedures and often do not provide rapid patient access to drugs. There is of course a balance to be struck between providing patients with rapid access to medicines and ensuring adequate information is available regarding the risk-benefit factors of the drug, sometimes termed the “evidence versus access” challenge (Eichler et al., 2015).
In recent times however, much has been done to try and improve the situation (Baird et al., 2014). In the EU, there are two means to fast track approval of a drug; Accelerated assessment and Conditional marketing authorisation.
The Conditional marketing authorisation process allows ‘the approval of a medicine that address unmet medical needs of patients on the basis of less comprehensive data than normally required. The available data must indicate that the medicine’s benefits outweigh its risks and the applicant should be in a position to provide the comprehensive clinical data in the future’ (EMA, 2015). In 2014, the EMA initiated a pilot project for a new model of drug testing and marketing called Adaptive Pathways (also known as Adaptive Licensing, or more recently the terms Medicines Adaptive Pathways’ (MAPs) or ‘Medicines Adaptive Pathways to Patients’ (MAPPs) have been used). This concept allows new drugs that would treat “unmet medical needs” to be launched on the market faster, based on an incomplete data set (Davis et al., 2016).
This project aims to discuss Adaptive Pathways in general, the different types of Adaptive Pathways, as well as the types of drugs that are suitable for these pathways. Finally, the Adaptive Pathway approval strategies in the EU and the US will be discussed.
2014 saw the introduction of a pilot program by which drugs could potential to make it to the market faster in order to fulfil unmet medical needs, the Adaptive Pathway. The idea was to ensure timely patient access to new drugs, while ensuring adequate risk benefit information was available. The Adaptive Pathways model presented by the EMA is not a new route of marketing authorisation but rather makes use of existing regulatory approaches such as the conditional marketing authorisation or the standard marketing authorisation. Using this pilot program, drugs could be authorized conditionally or in a staggered fashion using data gathered throughout the life of the product.
The three key principles of Adaptive Pathways are;
Bearing in mind that drugs are approved through adaptive pathways based on incomplete data and are given market approval earlier in the development process, iterative development refers to the gathering of data to increase knowledge after authorisation. It is a staggered approach to widen the target population or expand the indication. The data collected can also be used to reduce any uncertainties that were present at the early approval stage.
A key component of Adaptive Pathways is a well-defined prospective plan for collecting real-life data that can be used in conjunction with the Clinical Trials data to enhance the risk benefit ratio (Eichler et al., 2012).
Communication between stakeholders is critical and helps to decide what medicines are suitable, and are also responsible for creating an agreed prospective plan required for data gathering throughout the lifecycle of the product. Stakeholders are involved from a very early stage and can include health technology assessment (HTA) bodies, patients, as well as regulators and healthcare professionals.
The obvious advantage of the Adaptive Pathway is that the patient can potentially have access to a drug in a much shorter time period than if the drug had to go through the standard application process. If the drug shows a good risk benefit outcome, the drug can be approved at an earlier stage, while real-life data is gathered at predefined intervals from patients to confirm effectiveness. There is also the opportunity for drugs to be fully approved within a shorter timeframe than normal. Eichler also suggests that this process “may reduce the overall cost of development by allowing better-informed decisions on product viability to be made earlier in the development process” (Eichler et al., 2012). Eichler has published an article detailing the enablers of this new concept (Eichler et al., 2015).
However, since the publication of the EMA’s final report in July following the completion of the pilot program, a number of articles have been published which criticise the new concept (Eichler et al., 2012) (Woodcock, 2012). Unlike the standard authorisation, the Adaptive Pathway uses preliminary data and omits a number of steps that were designed to protect patients from unsafe and ineffective drugs and, this raises concerns as to whether this can potentially lead to increased risk to the patient.
For adaptive Pathways to work, it is critical that evidence obtained after initial approval be taken into consideration. However, it has been found that healthcare professionals are often slow to abandon unsafe methods (Tatsioni et al., 2007). This is worrying as the basis of Adaptive Pathways is that reliable data is generated after authorisation that will shed more light on the risk benefit to the patient.
Another serious concern relates to the life cycle management of the new drug (Davis et al., 2016). As mentioned earlier, a key principle of the Adaptive Pathway is gathering real-life evidence in post-marketing studies that would be used to update the risk benefit information. However, it would seem that in the case of conditionally approved drugs, the regulatory bodies have failed to ensure post-marketing study commitments are followed through (Banzi et al., 2015). It has also been suggested that where undesirable findings are encountered, companies may dispute these as unreliable results (McCabe et al., 2010) or may discount them because there are large financial gains or reputations at risk (Prasad et al., 2012).
Moreover, the willingness of stakeholders to participate in a program that involves more uncertainty, and correspondingly perhaps more risk, than before will be one of the greatest challenges to the Adaptive Pathway.
The report issued by the EMA suggests that drugs that treat infectious diseases, Alzheimers disease, degenerative diseases and rare cancers are potential candidates for this process with the objective of furthering their development and that suitable for the Adaptive Pathways would be those that treat rare diseases, where clinical data is not that common. The EMA have provided a flowchart to help companies determine if a product is an appropriate candidate or not, (Figure 2). Based on this, the key features of a drug that could be deemed suitable for this Adaptive Pathways approach are; iterative development, collaboration with HTA, and the use of real time data for regulatory purposes.
Figure 2 Adaptive Pathway product eligibility flowchart
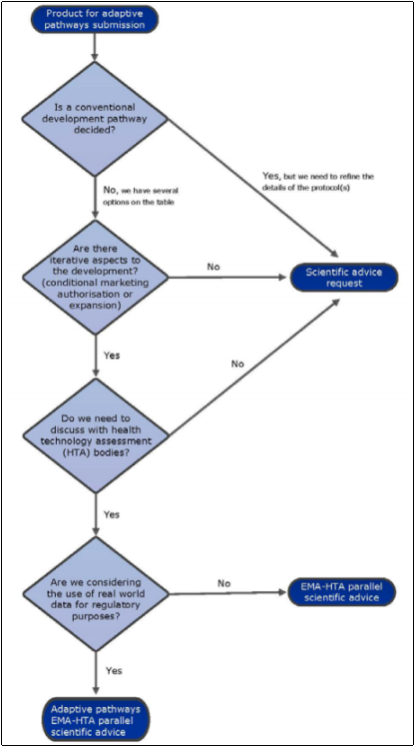
While there is no strict restriction on what type of drugs are eligible for the Adaptive Pathways, the EMA states that this route is not “to be applicable to all medicines, but only to medicines that are likely to address an unmet medical need”(EMA, 2016). During the pilot, 62 applications were received from a variety of therapeutic areas with cancer therapies accounting for a large proportion (33%). It is worth noting that Orphan designated drugs are suitable candidates, with 5 making it through to Stage 1 Meetings (15 drugs had been given Orphan drug designation by the European Commission at the time of submission) (EMA, 2016).
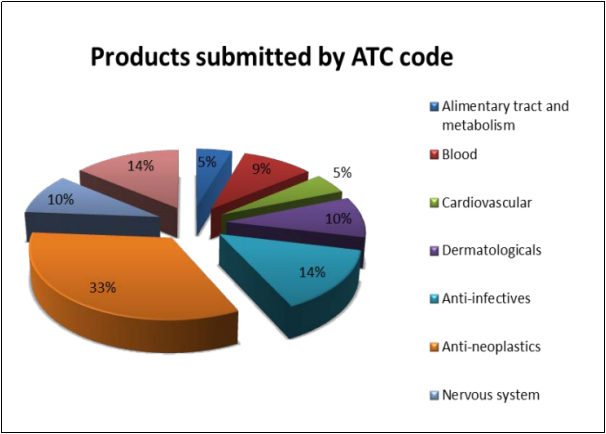
(EMA, 2016)
2.5. Adaptive Pathway approval strategy in the USA
Within the USA, there are four approaches to getting drugs on the market as rapidly as possible, and in each case the drug must be intended to treat a serious condition. A serious condition is defined as a disease or condition associated with morbidity that has a substantial impact on day-to-day functioning
Fast Track refers to the process, approved in 1992 under the Prescription Drug User Fee Act, by which drugs needed to treat serious conditions or to fulfil unmet medical needs are rapidly approved. According to the FDA, “filling an unmet medical need is defined as providing a therapy where none exists or providing a therapy which may be potentially better than available therapy” (FDA, 2014). This strategy means more communication with the FDA regarding the drug development plan, data collection and clinical trials design. In addition, a Fast Track designated drug is potentially eligible for Accelerated Approval and Priority Review if it meets the applicable criteria.
Breakthrough Therapy designation accelerates the development of drugs needed to treat serious conditions that have shown substantial advantages over existing treatments in early clinical studies (Poirier and Murphy, 2016). This strategy utilises a surrogate endpoint. A surrogate endpoint is a marker used to determine effectiveness of a drug, such as the shrinking of a tumour and is often used rather than actual clinical endpoints, such as survival rates. Another example would be a significantly improved safety profile compared to available therapy (FDA). In addition to the Fast Track designation benefits, Breakthrough Therapy drugs receive a vast amount of guidance with the drug development program.
References
BAIRD, L. G., BANKEN, R., EICHLER, H. G., KRISTENSEN, F. B., LEE, D. K., LIM, J. C. W., LIM, R., LONGSON, C., PEZALLA, E. & SALMONSON, T. 2014. Accelerated access to innovative medicines for patients in need. Clinical Pharmacology & Therapeutics, 96, 559-571.
BANZI, R., GERARDI, C. & GARATTINI, S. 2015. Approvals of drugs with uncertain benefit-risk profiles in Europe. European journal of internal medicine, 26, 572-584.
DAVIS, C., LEXCHIN, J., JEFFERSON, T., GØTZSCHE, P. & MCKEE, M. 2016. ” Adaptive pathways” to drug authorisation: adapting to industry? BMJ: British Medical Journal, 354.
EICHLER, H. G., BAIRD, L. G., BARKER, R., BLOECHLâ€DAUM, B., BØRLUMâ€KRISTENSEN, F., BROWN, J., CHUA, R., DEL SIGNORE, S., DUGAN, U. & FERGUSON, J. 2015. From adaptive licensing to adaptive pathways: Delivering a flexible lifeâ€span approach to bring new drugs to patients. Clinical Pharmacology & Therapeutics, 97, 234-246.
EICHLER, H. G., OYE, K., BAIRD, L. G., ABADIE, E., BROWN, J., L DRUM, C., FERGUSON, J., GARNER, S., HONIG, P. & HUKKELHOVEN, M. 2012. Adaptive licensing: taking the next step in the evolution of drug approval. Clinical pharmacology and therapeutics, 91, 426.
EMA. 2015. Fast track routes for medicines that address unmet medical needs [Online]. [Accessed].
EMA 2016. Final report on the adaptive pathways pilot.
FDA. Breakthrough Therapy [Online]. Available: http://www.fda.gov/ForPatients/Approvals/Fast/ucm405399.htm [Accessed].
FDA. 2014. Fast Track [Online]. Available: http://www.fda.gov/ForPatients/Approvals/Fast/ucm405399.htm [Accessed].
KASHYAP, U. N., GUPTA, V. & RAGHUNANDAN, H. V. 2013. Comparison of Drug Approval Process in United States & Europe. J Pharm Sci Res, 5, 131-6.
MCCABE, C., CHILCOTT, J., CLAXTON, K., TAPPENDEN, P., COOPER, C., ROBERTS, J., COOPER, N. & ABRAMS, K. 2010. Continuing the multiple sclerosis risk sharing scheme is unjustified. Bmj, 340, c1786.
POIRIER, A. F. & MURPHY, W. R. 2016. The Impact of Breakthrough Therapy Designation on Development Strategies and Timelines for Nononcology Drugs and Vaccines. Clinical Pharmacology & Therapeutics, 100, 603-605.
PRASAD, V., CIFU, A. & IOANNIDIS, J. P. A. 2012. Reversals of established medical practices: evidence to abandon ship. Jama, 307, 37-38.
RICK, N. 2009. Drugs from discovery to approval., John Wiley &
Sons, Inc.
TATSIONI, A., BONITSIS, N. G. & IOANNIDIS, J. P. A. 2007. Persistence of contradicted claims in the literature. Jama, 298, 2517-2526.
WOODCOCK, J. 2012. Evidence vs. Access: Can Twentyâ€Firstâ€Century Drug Regulation Refine the Tradeoffs? Clinical Pharmacology & Therapeutics, 91, 378-380.
staggered or conditional marketing authorization
AL is expected to involve a trade-off between earlier access for some patients vs. an increased level of acceptable uncertainty about benefits and risks, although the degree of uncertainty is expected to diminish with additional evidence generation.
One of the main purposes of the AL scheme is to get more robust and more relevant data earlier and throughout product development. Any attempt to move toward a more adaptive approach would have to be complemented by appropriate communications to key stakeholders and assurance that the appropriate post-initial authorization capabilities exist for ongoing monitoring of medical products for which AL has been applied
You have to be 100% sure of the quality of your product to give a money-back guarantee. This describes us perfectly. Make sure that this guarantee is totally transparent.
Read moreEach paper is composed from scratch, according to your instructions. It is then checked by our plagiarism-detection software. There is no gap where plagiarism could squeeze in.
Read moreThanks to our free revisions, there is no way for you to be unsatisfied. We will work on your paper until you are completely happy with the result.
Read moreYour email is safe, as we store it according to international data protection rules. Your bank details are secure, as we use only reliable payment systems.
Read moreBy sending us your money, you buy the service we provide. Check out our terms and conditions if you prefer business talks to be laid out in official language.
Read more