Janis Szeremeta
Epigenetic control of endocannabinoid function
Prostate cancer is one of the most frequently diagnosed types of tumours in the male population worldwide. The endocannabinoid system, more specifically high expression of cannabinoid receptor 1 (CB1) in tumour tissue, has been associated with poor prognosis in prostate cancer and suggested as a prognostic marker. Epigenetic silencing has previously been shown to upregulate CB1 mRNA expression in colon cancer cell lines and to induce expression of normally silenced cannabinoid receptor 2 (CB2) mRNA in a neuroblastoma cell line. In the present study, potential effects of epigenetic modulation on the expression of 12 different components of the endocannabinoid system (receptors, synthetic and catabolic enzymes) were investigated in a prostate cancer and a neuroblastoma cell line. Additionally, two catabolic pathways were investigated in functional assays. In general, changes in mRNA expression levels produced by treatment with the epigenetic modulators, 5-aza-2′-deoxycytidine and Trichostatin A were small, and, in the case of the catabolic enzyme fatty acid amide hydrolase in DU-145 prostate cancer cells were not accompanied by observable changes in hydrolysis rates. In SH-SY5Y neuroblastoma cells a low expression of monoacylglycerol lipase was found and this was also observed in functional assays. It is concluded that for the cell lines investigated, the epigenetic modulators tested do not modify the endocannabinoid system to any obvious degree, at least at the mRNA level. Since these experiments were conducted on a single cell line of a specific cell type only, introduction of alternative prostate cancer cell lines, such as PC-3 or LNCaP, might have different outcomes and should be considered for future experiments.
Due to its involvement in a variety of physiological and pathophysiological conditions, such as obesity, pain, immunomodulation and cancer1, the endocannabinoid system has emerged as an important area of research. Endogenous lipid transmitters, the so-called endocannabinoids, act by binding and activating the G-protein coupled cannabinoid receptors 1 and 2 (CB1/ CB2). Endocannabinoid levels are tightly regulated by a network of synthesizing and catabolizing enzymes (Figure 1). Two lipid mediators, N-arachidonoylethanolamine (anandamide, AEA) and 2-arachidonoylglycerol (2-AG), remain the most thoroughly studied endocannabinoids to date. 2-AG is derived from hydrolysis of diacylglycerols (DAGs) containing arachidonic acid via diacylglycerol lipases α and β (DGLα/β) and then hydrolysed to arachidonic acid mainly via monoacylglycerol lipase (MGL) but also by α/β-hydrolase domain containing 6 and 12 (ABHD6, ABHD12)2. AEA is derived from N-acylphosphatidylethanolamines (NAPEs) by hydrolysis via NAPE-phospholipase D (NAPE-PLD). It is inactivated by hydrolysis via fatty acid amide hydrolase (FAAH) and N-acylethanolamine acid amide hydrolase (NAAA) to arachidonic acid. Arachidonic acid is a substrate for many enzymes, including cyclooxygenase (COX) -1 and -2, 5- and 12-lipoxygenases (5/12-LOX) to produce prostaglandins, 5- and 12- hydroxyicosatetraenoic acid (5/12-HETE), respectively. Both 2-AG and AEA can also be hydrolysed to prostaglandin H2 derivatives via COX-23. Current modulators of the endocannabinoid system include a variety of selective pharmacological inhibitors for these enzymes which can be used to study their functional roles in the body (see Figure 1 for compounds used in this study).
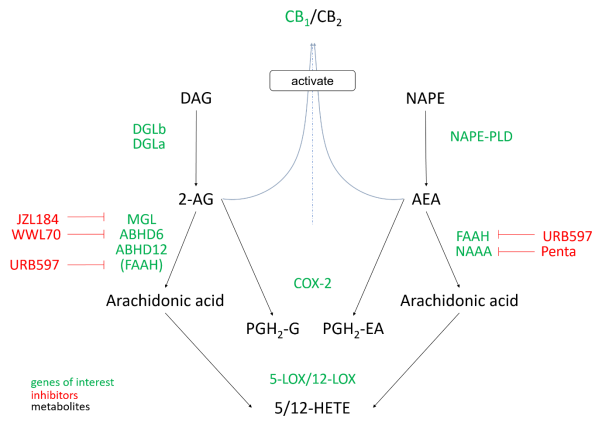
Figure 1: Simplified view of the endocannabinoid system. G-protein coupled receptors CB1 and CB2 are activated by lipid mediators, in this case 2-AG and anandamide (AEA) as well as by plant derived and synthetic compounds (not depicted). 2-AG and AEA are synthesized from diacylglycerol or N-acylphosphatidylethanolamine precursors and act locally. Both messengers are hydrolysed to arachidonic acid and/or prostaglandin H2 derivatives. Descriptions given in green were investigated towards changes in mRNA expression following epigenetic modulation treatment. Descriptions given in red show endocannabinoid metabolizing enzyme inhibitors. Abbreviations: Penta, Pentadecylamine (after Muccioli 20103).
The endocannabinoid system is becoming a more and more important therapeutic target in cancer, and very interestingly, different types of cancer appear to react differently to changes in endocannabinoid balance, with oftentimes opposing effects ranging for example from pro- to antiapoptotic4. This shows why understanding how the endocannabinoid system is regulated in health and disease remains an important part of research.
An important hallmark of cancer formation of cancer is the occurrence of epigenetic alterations5,6. Aberrant DNA methylation has been found in various types of cancer and effects vary between hyper- and hypomethylation states and in different types of cancer (see Kulis et al 20107). DNA methylation is usually associated with inhibition of gene expression. Cytosine nucleotides are methylated at the fifth carbon to form 5-methylcytosine, which can hinder transcription factor binding and therefore interfere with gene expression8. 5-Aza-2′-deoxycytidine is a DNA demethylation compound that is able to replace and mimic cytosine in the DNA. In case of a cytosine replacement, DNA methyltransferases (DNMTs), that would normally catalyse methylation of cytosines, will now be bound covalently to 5-Aza-2′-deoxycytidine, leading to degradation and depletion of DNMT protein levels and therefore a decrease of DNA methylation9. Note that this process is unspecific and generally decreases overall DNA methylation. Histone acetylation, a different type of epigenetic modification, is associated with activation of gene transcription. Occurring on lysine residues of histones, histone acetylation is associated with a charge neutralization of the positively charged histone molecules. This neutralization reaction is thought to decrease interaction between negatively charged DNA phosphate backbones and their positively charged histone counterparts, therefore increasing DNA availability10. Histone acetylation is regulated by an interplay of histone acetylases (HATs) and histone deacetylases (HDACs)11. Inhibition of HDACs may be used to constitutively activate histone acetylation mediated gene expression.
Prostate cancer has become one of the most frequently diagnosed malignancies in men throughout Europe12. Current evidence suggests that high a CB1 receptor immunoreactivity is correlated to disease severity and outcome13. Several prostate cancer cell lines and human prostate cancer tissues have been shown to express CB1 receptors using various techniques, such as qPCR, immunofluorescence and western blotting13-16. There is evidence that CB1 expression is regulated epigenetically in colorectal cancer, where DNA hypermethylation lead to a loss of CB1 expression17. The same study found inhibition of epigenetic silencing (i.e. removal of DNA methylation) increased Cnr1 mRNA expression in seven out of eight colorectal cancer cell lines. A different study investigated the effects of two different epigenetic modulators, 5-Aza-2′-deoxycytidine (Aza dC) and Trichostatin A (TSA), a histone deacetylase inhibitor, upon CB receptor expression in two different cell lines18. Inhibition of epigenetic silencing in Jurkat T cells increased Cnr1 mRNA expression in an additive manner but did not affect Cnr2 mRNA expression, whereas treatment of human SH-SY5Y neuroblastoma cells lead to induction of normally silenced Cnr2 mRNA expression, again in an additive manner, but no changes in Cnr1 mRNA.
Whilst the above data implicate epigenetic regulation of CB receptors, it is not known whether it is seen in prostate cancer cells, and there is no data concerning the endocannabinoid synthetic and catabolic enzymes. In consequence, the present study investigated the effects of Aza dC and Trichostatin A treatment upon mRNA expression for 12 different endocannabinoid-related genes (see Figure 1). Differences that were found were investigated in hydrolysis experiments and changes in either AEA or 2-AG hydrolysis. In addition, since tumours are often located in hypoxic microenvironments19, cell lines were exposed to hypoxic conditions for increasing intervals up to 24 h and the same panel of endocannabinoid system components was investigated towards mRNA expression. Cells were either placed into anoxic incubation chambers or exposed to hypoxia mimetics such as Co(II)Cl220 or deferoxamine21.
Drugs and Compounds
Radiolabeled compounds ([3H]-2-OG (60 Ci/mmol)), [3H]-AEA (60 Ci/mmol)) were obtained from American Radiolabeled Chemicals Inc, St. Louis, MO, USA. URB597, JZL184, WWL70 were obtained from the Cayman Chemical Co. (Ann Arbor, MI, USA). Pentadecylamine, 5-Aza-2′-deoxycytidine (Aza dC), Trichostatin A, Co(II)Cl2 were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Cell Culture
Human DU-145 (prostate cancer, passage range 17 to 29) and SH-SY5Y (neuroblastoma, passage range 19 to 28) cells were expanded in Eagle’s Minimal Essential Medium (EMEM – ATCC 30-2003) supplemented with penicillin, streptomycin (10,000 U/mL each, Gibco by Life Technologies) and 10% FBS (Gibco by Life Technologies) in 75 mL flasks at 37ËšC with 5% atmospheric CO2. Cells were plated in 24 well plates with a total number of cells of 1.5 Ã- 105 for DU-145 and 2.5 Ã- 105 cells for SH-SY5Y per well overnight.
Epigenetic Modulation using 5-Aza-2′-deoxycytidine and Trichostatin A
Following the overnight plating, DU-145 and SH-SY5Y cells were treated by replacing the old medium with a fresh layer of medium containing Aza dC (1 µM), Trichostatin A (25 nm), a combination of both, or vehicle (DMSO 0.1%) as control for 24 h. After 24 h hours, cells were lysed according to the Dynabeads® mRNA DIRECT„¢ Purification Kit (Thermo Fisher Scientific, Waltham, MA, USA) instructions and mRNA was extracted.
Exposure to Hypoxia/Hypoxia Mimetics
Induction of hypoxia was achieved via two different methods. Cells were seeded into 24 well plates and either kept in a hypoxic environment or were exposed to the hypoxia mimetic Co(II)Cl2. A hypoxic atmosphere inside an airtight modular incubation chamber (Billups Rothenberg Inc, San Diego, CA, USA) was achieved by first flushing the medium with a hypoxic gas mix (1% O2, 99% CO2) at a rate of 3 L/min for 5 minutes. The old medium was replaced with a layer of flushed medium and plates were placed into the airtight chamber. The chamber was flushed with hypoxic gas at a rate of 20 L/min for 5 minutes (per manufacturers’ instructions22) and then incubated at 37ËšC for either 2, 4, 6, 8 or 24 h. Co(II)Cl2 was used at a final concentration of 50 mM and cells were incubated for 2, 4, 6, 8 or 24 h. HIF1α and HIF2α mRNA levels were assessed for both procedures to evaluate induction of hypoxia.
qPCR
mRNA was extracted using the Dynabeads® mRNA DIRECT„¢ Purification Kit. mRNA (5 µg of total) was used for reverse transcription using the High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Applied Biosystems, Thermo Fisher Scientific). qPCR reaction mixtures were prepared using the KAPA SYBR FAST qPCR Master Mix (2X, KAPA Biosystems, Wilmington, MA, USA) to a final Volume of 20 µL. Reactions were run on the Illumina Eco Real Time PCR system (Illumina Inc, San Diego, CA, USA) with an initial denaturation time of 10 minutes at 95ËšC, 45 cycles of 10 seconds at 95ËšC and 30 seconds at 60ËšC and melting curve cycle times of 15 seconds at 95ËšC, 15 seconds at 55ËšC and a final step of 95ËšC for an additional 15 seconds. Primers (Table 1) were synthesized at Integrated DNA Technologies (Coralville, IA, USA). Amounts of transcripts were normalized to ribosomal protein L19 (RPL19) and relative quantification was performed using the ˆ†ˆ†Ct method.
Table 1: primers used for qPCR experiments
|
Gene |
Product |
Forward primer (5′ to 3′) |
Reverse primer (5′ to 3′) |
|
Abhd6 |
ABHD6 |
GATGTCCGCATCCCTCATAAC |
CCAGCACCTGGTCTTGTTTC |
|
Abhd12 |
ABHD12 |
GGCAGAAAGCTCTATAGCATCG |
CCTGTAGCCAAGGTCTGAATG |
|
Cnr1 |
CB1 |
CACCTTCCGCACCATCACCAC |
GTCTCCCGCAGTCATCTTCTCTTG |
|
Cnr2 |
CB2 1st pair |
CATGGAGGAATGCTGGGTGAC |
GAGGAAGGCGATGAACAGGAG |
|
CB2 2nd pair |
AAACAACTGGGACTCCTC |
GTCTAGAAGGCTTTGGGTTG |
|
|
Ptgs2 |
COX-2 |
AGCAGGCAGATGAAATACCAG |
ACCAGAAGGGCAGGATACA |
|
Dagla |
DAGLα |
CCCAAATGGCGGATCATCG |
GGCTGAGAGGGCTATAGTTAGG |
|
Daglb |
DAGLβ |
TCAGGTGCTACGCCTTCTC |
TCACACTGAGCCTGGGAATC |
|
Faah |
FAAH |
CACACGCTGGTTCCCTTCTT |
GGGTCCACGAAATCACCTTTGA |
|
Hif1a |
HIF1α |
GCTGATTTGTGAACCCATTCC |
TTCATATCCAGGCTGTGTCG |
|
Epas1 |
HIF2α |
CACAGAGTTCTTGGGAGCAG |
ACCCTTTGCAGACCTTGTC |
|
Alox5 |
5-LOX |
ATCCAGCTCAACCAAATCCC |
ACCAGATGTGTTCGCAGAAG |
|
Alox12 |
12-LOX |
GATCCGAGGAGAGAAGCAATAC |
GGAGGCTGAATCTGGATGAC |
|
Alox15 |
15-LOX |
CGAGGGTTTCCTGTCTCTTTAC |
GCACCCAAGAGTACCAGTC |
|
Mgll |
MAGL |
GGAAACAGGACCTGAAGACC |
ACTGTCCGTCTGCATTGAC |
|
Naaa |
NAAA |
ATGGAGCGTGGTTCCGAGTT |
AGGCTGAGGTTTGCTTGTCCT |
|
Napepld |
NAPE-PLD |
ACTGGTTATTGCCCTGCTTT |
AATCCTTACAGCTTCTTCTGGG |
|
Rpl19 |
RPL19 |
CACATCCACAAGCTGAAGGCA |
CTTGCGTGCTTCCTTGGTCT |
[3H]-AEA Hydrolysis in DU-145 Cells
The assay of Björklund et al. (2014)23 was used. Cells (1.5 Ã- 105 per well) were plated and kept overnight to allow for cell adherence. Subsequently, cells were treated with Aza dC (1 µM) for 24 h or left untreated as control. Non-enzymatic hydrolysis was measured in non-cell containing wells. Wells were washed with KRH buffer (120 mM NaCl, 4.7 mM KCl, 2.2 mM CaCl2.2H2O, 10 mM HEPES, 0.12 mM KH2PO4, 0.12 mM MgSO4 containing 1% BSA (Sigma Aldrich) followed by KRH buffer alone. KRH buffer containing 0.1% fatty-acid free BSA (Sigma Aldrich) was added to the wells and plates were kept in a water bath at 37ËšC. Inhibitors (URB597 1 µM, Pentadecylamine 1 µM, URB597 and Pentadecylamine 1µM each) or vehicle (DMSO 0.1%) were added and plates incubated for 10 minutes at 37ËšC. [3H]-AEA (diluted with non-radioactive AEA to give a final assay concentration of 0.5 µM) was added and plates were incubated for a further 15 minutes resulting in a total reaction volume of 400 µL. The hydrolysis reaction was stopped by adding 600 µL activated charcoal in 0.5 M hydrochloric acid and plates were kept on ice. Charcoal and aqueous phase were separated by centrifugation (2,500 rpm, 10 min.), 200 µL of the aqueous phase were recovered and mixed with 4 mL scintillation liquid (ULTIMA GOLD, PerkinElmer) for liquid scintillation radioactivity determination with quench correction. The [3H]-AEA used is labelled in the ethanolamine part of the molecule, and the [3H]-ethanolamine produced by the hydrolysis of [3H]-AEA does not adsorb to the charcoal, whereas the [3H]-AEA does adsorb24.
[3H]-2-OG Hydrolysis in SH-SY5Y Cells
Cells (2.5 Ã- 105 per well) were plated and incubated overnight to allow for cell adherence. Non-enzymatic hydrolysis was measured in non-cell containing wells. The assay used was the same as for [3H]-AEA hydrolysis, but using 0.5 µM [3H]-2-OG (labelled in the glycerol part of the molecule). Inhibitors (URB597 1 µM, JZL184 1 µM, WWL70 10 µM, a combination of URB597, JZL184 and WWL70 and a combination of JZL184 and WWL70 at the aforementioned concentrations) or vehicle (DMSO 0.1%) were added and plates incubated for 10 minutes at 37ËšC followed by addition of substrate and incubation for a further 15 min. See above for determination of radioactivity in aqueous phase.
Cytotoxicity Assessment/Assay
To determine the cytotoxicity of the various treatments throughout this project the LDH cytotoxicity detection kit from Roche (Cat. No. 11 644 793 001) was used per manufacturers’ protocol.
Statistical Analyses
Statistical analyses were undertaken by my Supervisor using the function ezANOVA in the package ez for the R statistical programme (R Core Team, URL http://www.R-project.org/). The details and the command lines used are given in Table 2.
Epigenetic regulation of endocannabinoid function
DU-145 and SH-SY5Y cells were treated for 24h with either Aza dC, TSA or a combination of both compounds, after which mRNA was extracted and analused for expression of marker of the endocannabinoid system.
Table 2 shows the summarized data of the statistical analysis obtained in the gene expression studies. Main effects are given in the left half of the table. Significant differences were found for a various number of genes and are given in bold type. “Main effects – cell” describes the comparison of gene expression between DU-145 and SH-SY5Y cells. The columns with Aza dC and TSA describe the effect of the epigenetic modulators on mRNA expression of the gene of interest and only a few of them were statistically significant (i.e. DGLβ and FAAH for Aza dC and 12-LOX for TSA).
Interpretation of the main effects is difficult when there are significant interactions. Values in bold type indicate an interaction between components) for four of the twelve genes of interest. In these cases, individual two-way ANOVAs helped to determine actual differences for each cell line per se. Results of these ANOVAs can be found below their corresponding figures (see Figure 2, Figure 3 and Figure 4) with a P<0.05 suggesting an effect of the epigenetic silencing treatment. Results will be discussed in more detail below.
Table 2: Three-way ANOVA summary for the PCR data.
|
Main effects |
Interactions |
||||||
|
Cell: |
|||||||
|
Cell: |
Cell: |
Aza dC: |
Aza dC: |
||||
|
Protein |
Cell |
Aza dC |
TSA |
Aza dC |
TSA |
TSA |
TSA |
|
CB1 |
0.0003 |
0.31 |
0.060 |
0.38 |
0.89 |
0.14 |
0.30 |
|
NAPE-PLD |
0.34 |
0.40 |
0.28 |
0.0093 |
0.29 |
0.29 |
0.54 |
|
DGLα |
<0.0001 |
0.87 |
0.88 |
0.0049 |
0.49 |
0.16 |
0.61 |
|
DGLβ |
0.43 |
0.0004 |
0.027 |
0.020 |
0.031 |
0.88 |
0.96 |
|
FAAH |
<0.0001 |
<0.0001 |
0.041 |
0.0061 |
0.55 |
0.17 |
0.85 |
|
NAAA |
<0.0001 |
0.012 |
0.53 |
0.44 |
0.79 |
0.15 |
0.40 |
|
MGL |
<0.0001 |
0.21 |
0.019 |
0.014 |
0.85 |
0.25 |
0.59 |
|
ABHD6 |
0.0004 |
0.019 |
0.15 |
0.0001 |
0.70 |
0.43 |
0.67 |
|
ABHD12 |
0.0078 |
0.014 |
0.65 |
0.091 |
0.14 |
0.61 |
0.11 |
|
COX2 |
<0.0001 |
0.032 |
0.62 |
0.21 |
0.70 |
0.83 |
0.74 |
|
5-LOX |
0.99 |
0.45 |
0.21 |
0.91 |
0.98 |
0.13 |
0.53 |
|
12-LOX |
0.0039 |
0.18 |
0.0001 |
0.41 |
0.55 |
0.93 |
0.69 |
Data shows the ANOVA p values for each protein, calculated for the data expressed as ˆ†Ct using the function ezANOVA in the package ez for the R statistical programme. The command line used was “Model<-ezANOVA(data=dataset, dv=.(Protein), wid=.(id), within=.(Aza dC,TSA), between=.(Cell), detailed=FALSE, type=3″ (Field, 201225). P values in bold type are those where significance remained after implementation of a 5% false discovery rate (Benjamini & Hochberg, 199526). When the interaction cell type x Aza dC was significant, two-way ANOVA matching for Aza dC and TSA have been calculated for each cell type separately, and these are shown in the figures. Note that for DGLβ and MGL the variances were different for the DU145 and SH-SY5Y cells and this will affect accuracy of the P values. In these cases, the cells have been analysed separately and the ANOVA values given in the figures.
Cannabinoid receptors 1 and 2
|
|
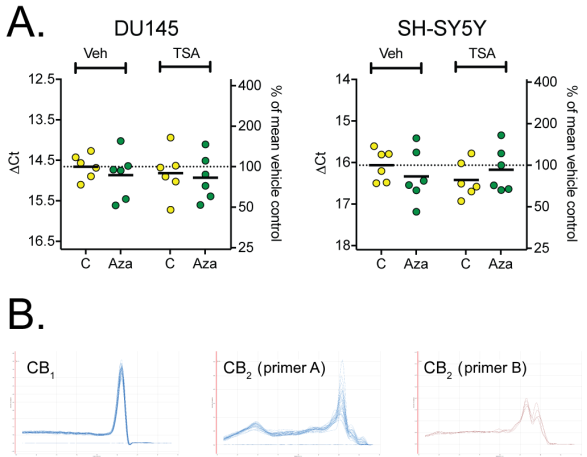
Figure 2: Panel A, mRNA levels for CB1 receptors in DU145 and SH-SY5Y cells treated with Aza dC and/or TSA. The graphs show the individual ˆ†Ct values (bars show the means), N=6 per group (each assayed in triplicate), with the corresponding % of controls on the right column. For statistical treatment, see Table 2. Panel B, melting curves for the primers used for CB1 and CB2 receptors. The melting curves are for the DU145 cells.
Gene expression analysis data of CB1 mRNA is given in Figure 2A. Expression rates were significantly different between the two cell lines, but neither Aza dC nor Trichostatin A had an effect. No interactions between the compounds and the cell types were found (Table 2)
Unfortunately, two different primer pairs, designed to amplify Cnr2 mRNA did not give detectable and reproducible mRNA expression of CB2, so no expression data could be obtained for CB2 (Figure 1B). The first primer pair was taken from a previous publication by Börner et al whereas the second pair was designed on site. Figure 1B shows the different melting curves obtained during the qPCR assays for DU-145, with similar results for SH-SY5Y cells.
Endocannabinoid synthetic enzymes
|
|
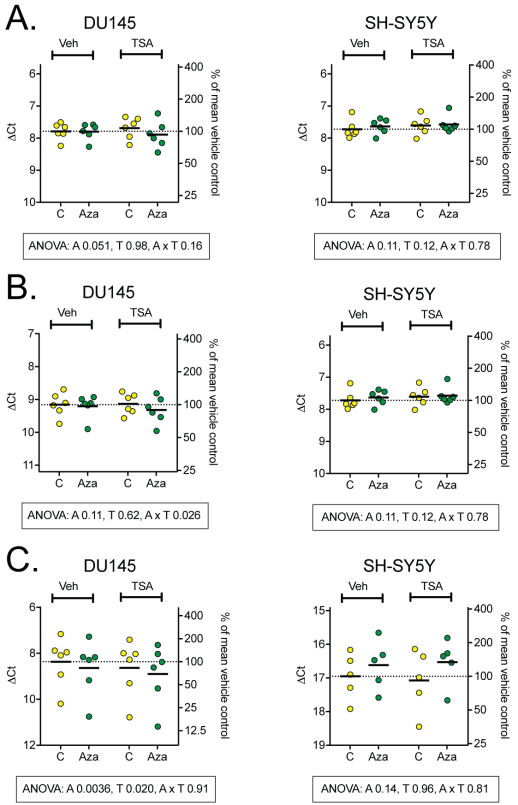
Figure 3: mRNA levels of the endocannabinoid synthetic enzymes NAPE-PLD (A), DGLα (B) and DGLβ (C). Two-way repeated ANOVA are shown when the interaction Cell x Aza dC in Table 2 was significant (Panels A and B) or when the variance was different for the two cell types (Panel C).
Effects of epigenetic modulation on the expression of endocannabinoid synthetic enzymes are shown in Figure 2. No main effects of either Aza dC or TSA were detected for NAPE-PLD or DGLα, there was an interaction between the different cell types and the Aza dC treatment, however (see Table 2). For these samples a two-way ANOVA was calculated and values are given below each figure. Indiviual treatments did not have any significant effect on the expression of both NAPE-PLD and DGLα (Figure 2A and B), an additive effect of Aza dC and TSA could be observed for the expression of DGLα in DU-145 cells, where expression decreased to a small degree. For DGLβ, since the variance was different for both cell types, a two-way ANOVA was calculated for each. No significant effects were observed for DGLβ expression in SH-SY5Y cells. However, both Aza dC and TSA had significant main effects in the DU-145 cells, although the sizes of the changes produced by the compounds were very small (Figure 2C).
AEA catabolic enzymes
|
|
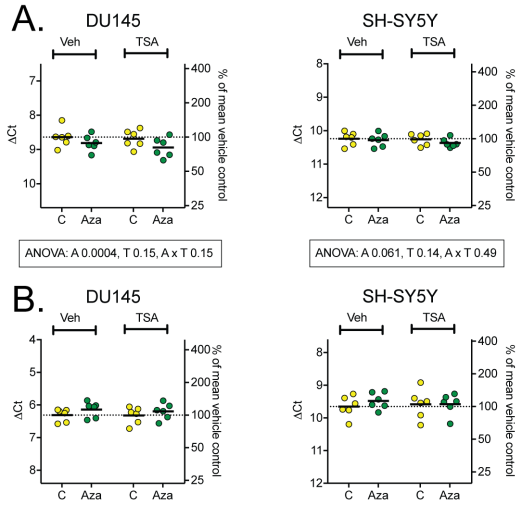
Figure 4: mRNA levels of the endocannabinoid catabolic enzymes FAAH (A) and NAAA (B). Two-way repeated ANOVA are shown when the interaction Cell x Aza dC in Table 2 was significant (Panel A).
As seen in Table 2, Aza dC had both a significant main effect, but also displayed interaction between the cell types and the compound for FAAH. The two-way ANOVA for FAAH resulted in significant differences only for the Aza dC treatment in DU-145, but not in SH-SY5Y. Once again, the effects were very small in size. Trichsotatin A did not have an effect in either cell line, neither individually nor in combination (Figure 3A). No significant differences were found for NAAA (Figure 3B).
2-AG catabolic enzymes
|
|
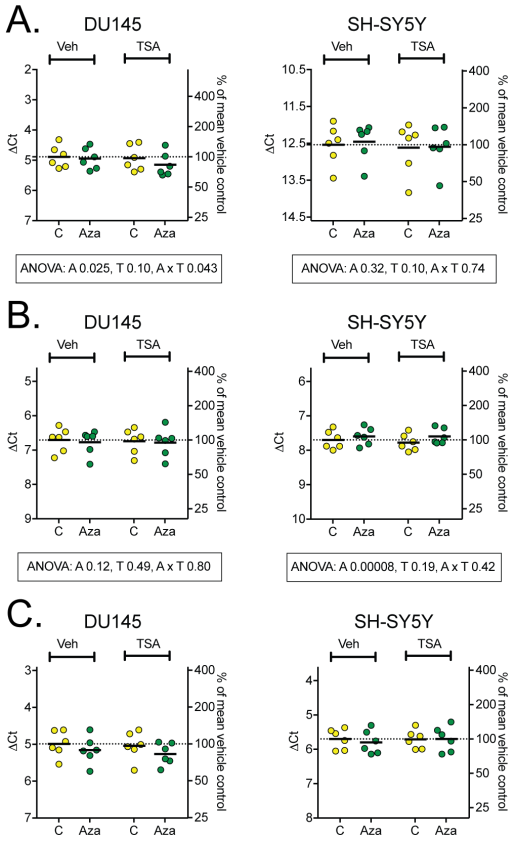
Figure 5: mRNA levels of the endocannabinoid catabolic enzymes MGL (A), ABHD6 (B) and ABHD12 (C). Two-way repeated ANOVA are shown when the interaction Cell x Aza dC in Table 2 was significant (Panel B) or when the variance was different for the two cell types (Panel A).
Gene expression analysis of the three key enzym
You have to be 100% sure of the quality of your product to give a money-back guarantee. This describes us perfectly. Make sure that this guarantee is totally transparent.
Read moreEach paper is composed from scratch, according to your instructions. It is then checked by our plagiarism-detection software. There is no gap where plagiarism could squeeze in.
Read moreThanks to our free revisions, there is no way for you to be unsatisfied. We will work on your paper until you are completely happy with the result.
Read moreYour email is safe, as we store it according to international data protection rules. Your bank details are secure, as we use only reliable payment systems.
Read moreBy sending us your money, you buy the service we provide. Check out our terms and conditions if you prefer business talks to be laid out in official language.
Read more