Glioma
Gliomas are the most common primary adult-onset malignant intracranial tumours. The world health organisation (WHO) classifies gliomas based on histological features indicative of prognosis. Low grade lesions (e.g. Grade I pilocytic astrocytoma) have a low proliferative capacity and are likely to be cured following tumour resection. Grade II lesions, which include diffuse astrocytomas and oligodendrogliomas, infiltrate normal brain parenchyma and have potential to develop into higher grade malignancies, in spite of their low proliferative capacity. Grade III are malignant lesions that are anaplastic and display mitotic activity (anaplastic astrocytoma). Additionally, tumours disaying microvascular proliferation with or without necrosis are termed glioblastoma (grade IV astrocytoma, or GBM)(Louis et al., 2007). Malignant or high grade (grade III-IV) gliomas represent a high proportion of brain tumour diagnoses. In particular, glioblastoma, the most lethal CNS tumour accounts for 54% of all glioma cases (Ostrom et al., 2016). Although global incidence rates are low (5.3 in 100,000), the highly invasive nature of these tumours results in high morbidity and mortality rates (ref). Currently therapeutic interventions against GBM include maximal surgical resection, radiotherapy and/or Temozolomide chemotherapy (Stupp et al., 2005). Furthermore, interventions fail to remove entire tumour mass resulting in recurrent disease and treatment resistance in tumour cells left behind. The post-treatment survival outcome, with standard-of-care therapy, is 12-16 months for glioblastoma and 2-5 years for anaplastic astrocytoma. The challenges to the development of efficacious drugs are a consequently affected by the heterogeneity of GBMs. In fact, the effectiveness and survival benefit of Temozolomide, a DNA alkylating agent which is the primary chemotherapy drug used for glioma patients, is correlated with tumour MGMT promoter methylation status (Hegi et al., 2005).
Aetiology and epidemiology
There are few established contributing risk factors to the development of gliomas. Exposure to ionising radiation is the only conclusive factor that increases the likelihood of glioma formation. Although familial patterns of glioma are rare, heritable variants carrying higher risk are associated with rare genetic syndromes such as neurofibromatosis types 1 and 2, the Li−Fraumeni syndrome (germ-line p53 mutations associated with an increased risk of several cancers including glioma), and Turcot’s syndrome (intestinal polyposis and brain tumours). (Malmer et al., 2007). Other genetic features associated with predisposition for glioma include certain variants of the EGFR receptor, TERT, CDKN2B in astrocytomas and CCDC26 in oligodendrogliomas. In the US, epidemiological studies report a trend of higher incidence within certain groups. For instance, malignant glioma is more prevalent in males who are 1.6 more likely to develop GBM than their female counterparts (Ostrom et al., 2016). Malignant gliomas are an age associated disease. The occurrence of anaplastic astrocytoma and GBM increase with age, and peaks at around 75-84 year group (Ostrom et al., 2016).
Patients with glioma often suffer from neurological deficits associated with tumour mass, increased intracranial pressure, surgical intervention and treatment response. Tumour associated epilepsy is a common manifestation in glioma patients albeit more so in low grade gliomas. Around 30% of patients present with epileptic seizures which require further intervention with antiepileptic drugs (Rajneesh and Binder, 2009).
Heterogeneity in Glioblastoma
Malignant gliomas are a highly heterogeneous classification of tumours. For this section, the focus will be on glioblastoma which is most frequently diagnosed. Genomic expression studies carried out by The Cancer Genome Atlas (TGCA) and other groups have defined four distinct subtypes of GBM. There are three core pathways (Receptor tyrosine kinase signalling, P53 and RB tumour suppressor pathways) forming an interconnected network of genetic abnormalities which drive tumourigenesis of GBMs. Analysis of the gene expression patterns of GBM identified frequent somatic mutations in TP53, PTEN, NF1, EGFR, ERBB2, RB1 and PI3K2A genes (Verhaak et al., 2010). The pattern of mutations in GBM seemed to follow amplification of oncogenic signalling pathways and deletions in tumour suppressor pathways(Brennan et al., 2014). Their analysis of these subclasses revealed four robust clusters of gene expression which were termed the classical, mesenychmal, proneural and neural subtypes of GBM (Verhaak et al., 2010). The classical subtype was characterised by amplification in EGFR not frequently observed in the other subclasses. There was also a significant absence of P53 loss even though these are the most frequent GBM-associated mutations. Mesenchymal subtype was categorised based on the deletions and low expression of NF1, frequent mutations in PTEN and P53 (a negative regulator of RAS GTPase) and the presence of mesenchymal markers CHI3LI and MET(Phillips et al., 2006). The two major features of the proneural subgroup are the simultaneous focal amplification in addition to highly expressed PGFRA and mutations in IDH1 which are virtually exclusive to this group. This subtype is more observed in younger patients and also in secondary GBMS. Furthermore, IDH1 mutations correlates with a better prognostic outcome (Verhaak et al., 2010). There were no immediate exclusive genetic alterations across the neural subtype. However, they were distinguished due to the expression of neural cell markers (NEFL, GABRA1, SYT1 and SLC12A5), despite their morphological and histological similarity to other gliomas and normal astrocytes. Varied response to treatment in histologically identical tumours is often reported in GBM. The WHO have consolidated histological classification of gliomas with prognostic molecular features in the attempt to group tumours that are likely to respond similarly to particularly treatment (Louis et al., 2016). The characterisation of differential molecular patterns and deregulated oncogenic and tumour suppressive pathways within GBM is informative of distinct tumour genotypes that may be of use in guiding development of novel therapeutics.
Altered metabolism and the Warburg effect in glioma
Tumourigenesis is a multistep process requiring cumulative mutations that eventually lead to transformation of normal cells into oncogenic derivatives. In the clonal evolution of cells, each successive alteration provides a survival advantage which progressively leads to transformation. Consequences of alterations result in traits which allow cancer cells to operate outside usual cellular regulatory networks. Hanahan and Weinberg described the classical hallmarks of cancer as the acquirement of self-sufficient growth signalling that is independent of exogenous growth factor stimulation, evasion of apoptosis, insensitivity to anti-growth factor signals, angiogenesis, invasion and metastases and having unlimited replicative potential as a result of environmental uncoupling leading to cell autonomy (Hanahan and Weinberg, 2000). Metabolic reprogramming is now a well-established characteristic of cancer cells (Hanahan and Weinberg, 2011). Alterations in cellular energetics provides supportive mechanisms for aberrant growth, survival and invasion in a harsh tumour microenvironment (Heiden et al., 2009). The most commonly described metabolic abnormality in cancer cells is the Warburg effect, a phenomema where cancer cells decouple glycolysis from pyruvate oxidation even periods of oxygen abundance. Under normal physiological conditions, oxidative phosphorylation is the dominant pathway for ATP production. This metabolic switch was initially puzzling due to OXPHOS producing significantly larger quantities of ATP (36 moles of ATP per glucose molecule) compared to aerobic glycolysis with results in net 6 moles of ATP per glucose molecule. The question remains as to why cancer cells exhibit this metabolic phenotype when it is the inefficient mechanism for ATP generation? Cancer cells experience a much harsher microenvironment than normal cells, with fluctuations in nutrient availability and oxygen due to abnormal vasculature. Although glycolysis is inefficient, it has a fast turnover for ATP production and is not limited by oxygen concentrations which is beneficial for maintaining proliferation and survival of cancer cells. Additionally, lactate which is the by-product of aerobic glycolsis can be extruded into the extracellular milieu causing acidosis which can promote tumour cell infiltration into surrounding tissue.
Numerous studies have observed increased glycolysis as a feature of glioma (Oudard et al., 1996; Zhou et al., 2011) . The use of FDG-PET which is an imaging modality dependent on the increased cancer cell uptake of glucose remains controversial for gliomas. Although certain reports have shown correlation of FDG uptake and increasing grade, there are still some limitations in distinguishing gliomas from normal brain tissue. Furthermore approximately 30% of gliomas are FDG-PET negative display avidity for FDG-PET making tumour detection by this method unsuitable (Chen et al., 2005). It is often speculated that the increased glucose uptake is indicative of upregulation of glycolysis and ATP generation through this pathway. GBMs display high diverse inter- and intratumoural heterogeneity which is likely to impact of metabolic phenotypes. GBMs have distinct cell populations which display different metabolic phenotypes. For instance, cancer stem cell populations in GBM are often characterised by a higher oxidative capacity and are the most chemo and radio-resistant populations within a tumour(Vlashi et al., 2011). In both immortalised and primary cultured cell lines, ATP production is not wholly accounted for by glycolysis which suggests that other metabolic pathways contribute to energy production. Warburg’s observation of increased aerobic glycolysis whilst still relevant does not capture the metabolic complexity of cancer cells. Several studies have observed the contribution of various metabolic fuels activation of to the ATP turnover of cancer cells (Guppy et al., 2002; Bonnet et al., 2007; Michelakis et al., 2010). Altered metabolic reprogramming serves ATP production as well as the biosynthetic requirements of cancer cells. Cancer cells utilise a diverse range of substrates to supply fuel (ATP) and synthesis of macromolecules (Nucleic acids and lipids) which sustains aberrant proliferation and growth (DeBerardinis et al., 2008).
Fatty acid metabolism
While much of cancer cell metabolism research has concentrated on glucose utilization, other substrates, such as fatty acids, are increasingly being appreciated as major contributors to cancer cell bioenergetics. In fact, glioma cells engage in the beta-oxidation of fatty acids and de novo fatty acid synthesis. This metabolic dependency may be exploited for clinical neuro-oncology, particularly biomarker discovery and drug development. Such novel approaches may give new promise to these patients, who currently have few therapeutic options.

Figure 1. Schematic of Fatty acids A) Palmitate B)Linoleic acid
Fatty acids encompass a diverse range of metabolites with different FA chain lengths, number and positioning of carbon double bonds. Fatty acids not only contribute to energy generation through mitochondrial and peroxisomal β-oxidation, thereby supplying the citric acid cycle and the electron transport chain. They are critical components of anabolism within a cell, forming phospholipids which impact on membrane structure and dynamics and glycerophospolipids which act as signal transducers; generate autocrine signalling through active lipid molecules such as sphingosine-1-phosphate, lysophosphatidic acid, second messengers DAG,IP3 and PIP3; generation of paracrine signalling molecules like eicosenoids and endocannabinoids; and act as cofactors to FA chaperones (FABPs) which are necessary for the sequestering of lipid into droplets in response to environmental changes (e.g.hypoxia). Additionally, fatty acids can form post-translational moieties (e.g. palmitoylation) on growth factor receptors which mediates their localisation to the plasma membrane. While the role of fatty acids have been characterised in other cancers, it not clear the impact of these processes in glioma and its progression.
Furthermore, FAs can act as metabolic fuel substrates where their degradation through FA-β-oxidation (FAO) provides significant supply of ATP, NADH, FADH2 and NADPH. The relevance of FAO in cancer until recently had remained largely unexplored unlike other metabolic pathways such as glycolysis, glutaminolysis and fatty acid synthesis. The relevance of FAO in cancer until recently had remained largely unexplored unlike other metabolic pathways such as glycolysis, glutaminolysis and fatty acid synthesis. Recent studies on FAO are beginning to shed light on the significance of FAO in cancer metabolism
Fatty acid oxidation in cancer
A crucial feature distinguishing healthy cells from cancerous cells is altered lipid metabolism. In many solid tumours, increased de novo FA synthesis and consumption is observed which provide ATP, reducing species (NADH and FADH2) which support energy generation in appropriate pathways. Alterations in FA metabolism not only co-opt tumour growth but can impact sensitivity to therapy. For instance, the increase in concentrations of saturated FAs leads to reduced permeability of chemotherapeutic drugs through membranes of prostate cancer cells. Gene expression studies have identified upregulations of enzymes involved in lipogenic and lipolytic pathways. FASN and ACC are overexpressed in several tumours and correlate with worse prognostic outcomes. Additionally, upregulation of FAO enzymes, namely the acyl-dehydrogenases correlate with worse patient outcomes. Although FAO is understudied in comparison to glycolysis, glutaminolysis and even FAS in tumour cells, there is increasing evidence of its importance in cancer cells.
CPT1 and ATP generation
FAO as a bionenergetic pathway has an immense capacity to produce large quantities of ATP to fulfil energy requirements of tumour cell growth. The inhibition of CPT1 by Etomoxir decreased ATP production in myc-driven triple negative breast cancer (TNBC) in vitro and additionally slowed tumour progression in PDX model of TNBC. FAO derived ATP may also support oncogenic signalling pathways. The use of transmitochondrial cybrids of TNBC revealed the phosphorylation and activation of Src is dependent on ATP generated from mitochondrial oxidative phosphorylation(Park et al., 2016). Genetic knockdown of CPT1 and CPT2, abolished src autophophorylation and inhibited metastasis. In some cancer cancers (prostate, B cell lymphoma and high grade-gliomas etc.), FAO is the dominant energy pathway and its impairment leads to a reduction in proliferation and survival due to ATP depletion.
FAO and redox homeostasis
The production of reducing species namely, NADH and FADH2 are important in the production of ATP. However, FAO also generates NADPH through the generation of acetyl-CoA. Citrate which is a product of the TCA cycle is exported into the cytosol where it is involved in NADPH producing reactions. GBM SF188 cells which are oxidative are susceptible to ETX treatment. Inhibition of FAO in these cells causes ATP depletion along with oxidative stress due to a reduction in NADPH and GSH content. Additionally, the LKB1-AMPK axis required to maintain redox balance. Cells with inactivated AMPK exhibit NADPH depletion, oxidative stress which triggers apoptosis (Pike et al., 2011).
CPT1 and apoptosis
Prostate cancer which is a slow growing tumour has been shown to be less reliant on aerobic glycolysis and uses lipids as its primary fuel source. The combination of Etomoxir and orlistat (FAS inhibitor) produces a synergistic effect resulting in decreased expression of androgen receptor, mTor signalling and caspase 3 activation (Schlaepfer et al., 2014). Furthermore, CPT1a knockdown reduces AK signalling and increases apoptosis.
The compound ST1326 which inhibits CPT1 results in growth arrest, mitochondrial damage and induces apoptosis and accumulation of lipids in leukaemic cells (Ricciardi et al., 2015). Inhibition of FAO by Etomoxir also sensitises human leukaemic cells to apoptosis induction by ABT-737, a molecule that causes the release of pro-apoptotic BCL-2 proteins like BAK (Samudio et al., 2010). Furthermore, FAO regulates the apoptotic response by directly impacting BAK-dependent mitochondrial permeability transition.
Fatty acid oxidation in glioma
Glioma cells contain FASN, and indeed the expression of this enzyme increases with tumour malignancy(Tao et al., 2013). Recent results in our lab have also demonstrated that glioma cells use fatty acids as a preferred substrate(Lin et al., 2016). Specifically, human glioma cells primary-cultured under serum-free conditions require fatty acid oxidation to maintain both respiratory and proliferative activity. Several emerging lines of evidence directly or indirectly validate our findings using completely independent methods – including results from clinical dietary interventions (ketogenic diet tested in 2 clinical trials, 1 pre-clinical trial by DL Rothman 2016, all showing non-promising results in slowing tumour growth but clinical trials have not been published yet), in vivo radiolabelling studies conducted in orthotopic mouse models of malignant glioma using patient-derived cells (Mashimo et al., 2014), and investigations into the mobilization of lipid droplets as energy sources in cultured glioma cells (Tennant Lab) These and other findings will almost certainly add to this incipient area of research in the coming months and years.
Sources of fatty acids
In cellular energetics, fatty acids are an important metabolic substrate for fuel supply and building blocks of macromolecules. The importance of FA catabolism is demonstrated in cardiac myocytes, which have high energetic demands and rely primarily on oxidation of FAs for ATP production. While the majority of FAO takes place in the mitochondria, a small proportion of FAO also occurs in peroxisomes. FAs can be obtained from plasma, storage organelles and synthesised de novo.
Extracellular uptake
Uptake of free plasma FAs is facilitated by fatty acid transport proteins (FATPs) which are highly conserved across species. FATPs are family of 6 related proteins with tissue specific expression and subcellular localisations. FATPs translocate to the PM and facilitate the uptake of exogenous FAs into the cytosol. FATP1 shows more ubiquitous expression whereas FATP4 expression is localised to the brain. Apart from mediating uptake of FAs across plasma membrane, FATPs possess intrinsic acyl-coA synthetase (ACS) activity which catalyse their esterification with CoA to generate acyl-CoA (Gimeno, 2007).
De novo synthesis of fatty acids
De novo fatty acid involves the activity of complex multifunctional enzyme ACC which carboxylates acetyl-coA to malonyl-coA .The fatty acid synthase (FASN) then mediates series of condensation reaction converting malonyl-coA to long chain saturated FAs including palmitate. Complex FAs are synthesised through the activity of elongase and desaturase enzymes. However, this not the case for most cancers as increased lipogenesis is frequently observed (Rohrig and Schulze, 2016).
The synthesis of FAs begins with the generation cytosolic acetyl-CoA from TCA derived citrate catalysed by ATP citrate lyase (ACLY). Acetyl-CoA carboxylase (ACC) catalyses the ATP-dependent carboxylation of acetyl-CoA to malonyl-CoA, the rate limiting step to FA biosynthesis. Biosynthesis of endogenous long chain FAs (e.g. saturated FA palmitate) is largely facilitated by fatty acid synthase (FASN). FASN is a complex multifunctional enzyme with 7 individual catalytic activities which catalyses the repeated condensation reaction of acetyl-CoA with malonyl-CoA as a carbon donor and NADPH as a reducing coenzyme. In normal cells and tissue, FASN contributes minimally to cellular lipid pools as most FAs available result from uptake from extracellular (dietary) sources. FASN is normally transcriptionally and translationally regulated to maintain low cellular levels. In the liver and other lipogenic tissues, FASN expression is regulated by hormonal changes (i.e. insulin, glucagon). Furthermore, FAs are produced from carbohydrates during periods of excess which can be esterified into triacylglycerols for storage and later undergoing FAO. Newly synthesised FAs can undergo further modifications leading the production of diverse FA species and prevention of lipotoxicity. An example of such modification is the introduction of double bonds in the acyl-chain of FAs. The generation of mono-unsaturated FAs is catalysed by desaturase enzymes such as stearoyl-CoA desaturases (SCD).
Tumour cells contrastingly overexpress FASN caused by aberrant growth factor stimulation, oncogene stimulation and loss of tumour suppressor activity (Menendez and Lupu, 2007). Labelling with 14C-glucose established that de novo synthesis accounted for a significant proportion of esterified FAs in several tumour types (Medes et al 1953). Exacerbated lipogenesis is a frequent observation in cancer cells. RNA interference and pharmacological inhibition of ACLY and ACC impinge cell proliferation in vitro and decreased tumourigenecity in vivo. Furthermore, Studies have report high expression of FASN in numerous tumours including breast, prostate and colon cancer. In glioma, FASN overexpression corresponds to the WHO tumour grade (Grube et al., 2014). Increased expression of lipogenic enzyme like FASN correlates with poorer prognosis suggesting that FAS plays an important supportive role in tumour development.
Lipid droplets
Lipid droplets are intracellular storage organelles that act as a reservoir for lipids. In periods of abundance, cytosolic lipids are converted into neutral lipids mainly triacylglycerols and sterol esters, then sequestered into LDs. LDs are composed of an amphipathic lipid monolayer embedded with LD associated proteins that enclose and exclude the hydrophobic lipid core from the aqueous cytosolic environment. The ratios of TAGs: sterol esters differ across cell types and are related to the lipid requirements of a cell type. For instance adipocytes LDs contain high amounts of TAGs which can be utilised for energy generation during periods of starvation. Essentially, the type of lipids sequestered into LDs match cell specific lipid requirements.
The primary function of these LD structures is their ability to store lipids, maintain cellular lipid homeostasis and avoid lipotoxocity. Their neutral lipid core provide a source of molecules that a critical components of chromatin structure, lipid ligands for nuclear receptors (e.g. PPARs); and fatty acid trafficking (Welte, 2015). LDs undergo hydrolysis to provide cells with essential lipid building blocks when nutrients are limiting. Hormone sensitive lipase (HSL) abd adipocyte triglyceride lipase (ATGL) are the two best known lipolytic enzymes.
Rambold et al show that FAs accumulate in LDs during fed state in mouse embryonic fibroblasts (MEFs). However when these MEFs are starved, LDs are able to localise and fuse with mitochondria and through the activity of cytoplasmic lipases, FAs are transferred to mitochondria to undergo FAO. LDs therefore play an important role in FA trafficking and facilitate oxidative metabolism and avoidance of FA toxicity. Herms et al confirms the interaction of LDs and mitochondria during starvation. Additionally, AMPK is activated and increased LD mobility along detyrosinated microtubules thereby increasing likelihood of LD and mitochondria contact (Herms et al., 2015).
In transgenic mice overexpressing MYC, the inhibition of this pathway results in growth inhibition and apoptosis. Consequently leading to LD accumulation as a result of mitochondrial dysfunction. LDs can therefore be a marker of cellular toxicity and treatment response. In GBM cells, cell survival under nutrient deprived conditions is highly dependent on LD derived FAs which undergo oxidation thus maintaining cell viability(Cabodevilla et al., 2013). The presence of LDs is a biomarker in GBM which may be indicative of tumour grade. For instance low LD accumulation is observed in low grade glioma. The presence of LD accumulation also correlates with poor prognosis (Geng et al., 2016).
Biochemical overview of Fatty acid oxidation
Carnitine pamiltoyl transferase shuttle
Acyl-coAs within the cytoplasm are impermeable to the mitochondrial outer membrane. Entry of acyl-coAs commits them to catabolism, hence, entry into mitochondria is a critical regulatory checkpoint and a major determinant of FAO. Carnitine Palmitoyl Transferase I (CPT1) sits on the outer mitochondrial membrane facing the cytoplasm where it catalyses the reaction converting acyl-coAs into acylcarnitines. Inside the mitochondria, Carnitine-Acylcarnitine Translocase (CACT) exchanges acylcarnitines for carnitines, ensuring relatively stable levels of carnitine in the cytosol to mitochondria. Carnitine Palmitoyl Transferase II (CPT2) sits on the inner mitochondrial membrane and is responsible for the reconversion of acylcarntine to acyl-coA which can then be catabolised. There exist three isoforms of the CPT1 enzyme, CPT1a, CPT1b and CPT1c, which show tissue-specific expression. CPT1a is the more widely expressed isoform whereas CPT1c expression is limited to the brain. The product of FA synthesis, malonyl-coA, binds to CPT1 and acts a physiological inhibitor of FAO (McGarry et al., 1978). As a result, fatty acid synthesis and oxidation can be temporally compartmentalised. CPT1a has the highest biding affinity to malonyl-coA making it the most prevailing isoform and the target for inhibition of FAO. Although CPT1c has sequence homology to the CPT1a and CPT1b isoforms; binds malonyl-CoA, it however does not possess acyl-transferase activity(Wolfgang et al., 2006). A metabolomics study found that there are only subtle changes in FAO metabolites in CPT1C knockouts, however increased levels of glutathione are observed (Lee and Wolfgang, 2012). CPT1C reportedly contributes to the metabolic transformation of cancer cells. Its upregulation results in strong activation of the mTor pathway in primary murine lung tumour models and contributes to rapamycin resistance (Zaugg et al., 2011). Additionally, its upregulation results in increased FAO, ATP generation in response to glucose deprivation and hypoxia. CPT1 knockdowns become more susceptible to metabolic stress as a result of decreased ATP production (Reilly and Mak, 2012). The contribution of CPT1C to FAO is still unclear however it may play a role in regulating response to metabolic stress.

FA Chain shortening by acyl-coA dehydrogenase and mitochondria trifunctional proteins
Once inside the mitochondrial matrix, acyl-CoAs can be freely oxidised. FAO is a cyclical process where acyl-coA chains are shortened. Each round results in the production of acetyl-coA released from the two carboxy-terminal carbons. These molecules, as well as the electron carriers FADH2 and NADH, are then directed to the TCA cycle and electron transport chain.FAO occurs in four principle steps-Dehydration– The acyl-CoA-ester undergoes dehydration catalysed the by acyl-CoA-dehyrogenases to trans-2-enoyl-coA. Hydration– Then hydration of the double bond to L-3-hydroxy-acyl-coA. Dehydrogenation– followed by dehydrogenated to 3-Keto-acyl-coA.Thiolytic cleavage– The final step involves a thiolytic cleavage producing acetyl CoA and an acyl-coA chain that is two carbons shorter. The resulting shorter acyl-CoA undergoes the cyclical process again until all carbons are liberated (Houten and Wanders, 2010).
Long chain FAs are shortened by membrane bound VLCAD (C12 to C24) and the mitochondrial trifunctional protein (encoded by HADHA or HADHB) which is a trienzymatic complex that possesses, hydratase, long chain hydroxyl-acyl-coA dehydrogenase and thiolase capability. After a few cyclical series, dehydrogenation is taken over by MCAD and finally by SCAD. Interestingly, steps 2-4 are carried out by distinct enzymes following dehydrogenation by MCAD (C4 to C12) or SCAD (C4 to C6). The medium and short chain hydroxyacyl-coA dehydroganse and medium chain 3-ketoacyl-coA thiolase have specificity for only ten carbon coA esters. Uneven number carbon acyl-coAs such as the monounsaturated linoleic acid require the activity of dodecenoyl-coA delta isomerase whereas polyunsaturated FAs need a second auxiliary enzyme 2, 4-dienoyl-coA for complete oxidation (Diedrich and Henschel, 1990). LCAD (C8 to C20) has very low expression in humans but plays an increased role in mouse FAO. The recently-discovered ACAD9 bears similarities to VLCAD however high levels in brain and displays optimal activity to palmitoyl-CoA (Zhang et al., 2002; Ensenauer et al., 2005).
Coupling of FAO to ETC
The products of β-oxidation and chain shortening are destined for the TCA and ETC where they contribute to ATP generation and anaplerosis respectively. The production ATP from FAO is through cofactor production and acetyl-coA. The function of the TCA is produce generate high energy electrons from metabolic fuel which in turn supply the ETC. The cycle sees entry of two carbon acetyl-coA and two carbons leave as CO2. It produces hydrides that are transferred to make 3 molecules of NADH and FADH2.Final common pathway for oxidised metabolites; carbohrydrates, lipids and amino acids playing a role in both catabolism and anabolism (generation of precursors for biosynthesis).The activity of the TCA itself does not produce ATP neither does it consumed oxygen. However it couples the oxidation of substrates to the ETC.
The mammalian respiratory chain involves protein complexes situated on the inner mitochondrial membrane that act as electron carriers. The movement of electrons from one complex to the next generates proton motive force that drives F1F0 ATPase.
The transport of electrons has two entry ports. Firstly via Complex I (NADH-ubiquinone oxioreductase) which is the entry point for electrons donated by NADH. Coenzyme Q also termed ubiquinone carries electrons from complex I to complex III. Complex II (succinate quinone oxioreductase) can additionally receive electrons fed from TCA intermediate succinate which acts an electron donor to FAD. The transfer of electron to coenzyme q in this system does not amount to significant increases in free energy so isn’t coupled to ATP synthesis at this stage. FADH2 derived electrons only yield free energy at complex III and IV. Complex III and complex IV (cytochrome c oxidase) uses cytochrome c to reduce oxygen which is the final electron acceptor in OXPHOS.
Fatty acids v glucose as metabolic substrates for ATP production
Fatty acids quantitatively produce more ATP in comparison with glucose.In particular, the saturated fatty acid palmitate produces 3 acetyl-coA per 6 carbons atoms whereas the 6 carbons of glucose together produce 2 acetyl-coA molecules. FAs thus an important source of energetic fuel for cells.
Transcriptional regulation
AMPK, the balance between synthesis and oxidation of fatty acids
AMPK is often referred to as the master regulator of cellular energy metabolism and exerts control of fatty acid metabolism. It’s a heterotrimeric structure made of the subunits (α catalytic, regulatory βγ subunits) and is highly sensitive to cellular levels of AMP (Hardie et al., 2012). In rising levels of AMP and ATP depletion, AMPK is reversibly phosphorylated on threonine 172 residue by an upstream kinases such as LKB1 and CAMKK resulting in phospho-AMPK which is the active conformation (Hardie and Alessi, 2013). In its activated state, AMPK enhances catabolic pathways that generate ATP whilst switching off ATP consuming pathways. During periods of cellular stress (substrate deprivation and hypoxia) where ATP is limiting, AMPK regulates the activities on various metabolic enzymes including ACC1 and ACC2 which commit acetyl-coA to FA synthesis or cholesterol synthesis. The malonyl-coA produced by the two ACC isoforms exert distinct effects on FA metabolism. Mouse knockout studies reveal that ACC1 produced malonyl-coA is utilised in FA synthesis whereas that generated from ACC2 exclusively regulates FAO. By exterting inhibition of these two enzymes, AMPK ensures de novo FA synthesis (ATP consuming) does not occur whilst also liberating CPT1 of the allosteric inhibition via malonyl-CoA production (Hardie and Pan, 2002). The regulatory activity of AMPK avoids improper activity through pathways that would perturb the metabolic homeostasis making it a critical regulatory checkpoint.
Glucose oxidation v FAO (Randle effect).
Catabolic pathways form part of a complex interconnected network of several metabolic pathways and do not exist in isolation. Several pathways have been identified in cancer cells which contribute to the overall cellular requirements.
The Randle effect describes the functional coupling of glucose oxidation and fatty acid oxidation. This crosstalk between two distinct pathways results in a dynamic biochemical mechanism which controls fuel selection and adapts substrate supply. Fatty acids and glucose display functional antagonism as substrates of oxidation.Inhibition of glucose by fatty acids contributes to the so called glucose sparing effect, sparing the gluconeogenic precursors, pyruvate and lactate. The interaction between the two metabolic substrates is highly complex, involving allosteric control by pathway specific substrates, expression of key enzymes (including pyruvate dehydrogenase (PDH) and pyruvate dehydrogenase kinase (PDK)) and finally by metabolic regulators such as AMPK and SREBP. Although the Randle effect is more clearly defined in other disease contexts ( i.e. type 2 diabetes, fatty acid syndrome etc), its role in cancer remains largely unexplored.
Rising concentrations of mitochondrial NADH/NAD+ and acetyl-coA/coA ratios when FAO is active inhibits glycolysis at several step including glucose uptake, PFK and most severely at the level of PDH. Due to the importance of PDH in metabolic fate, its activity is complex and tightly regulated. The products of PDH mediated reactions exert allosteric control over it, namely pyruvate or its analogue dichloroacetate. In addition, the E1 subunit of the PDH subunit which has the pyruvate decarboxylase activity can undergo post translation modification which alters covalent bonds and induce conformational change in the PDH complex. The enzymes responsible for these are the mitochondrial enzymes PDK and PDP which phospho-inactivate and dephosphorylate and activate, respectively. Therefore the activity of PDH is determined by the levels of PDK and PDP. PDH mouse knockdowns result in non-viable male pups. However, supplementation with a high fat diet promotes survival although defects in cardiac and skeletal muscle is observed at later developmental stages. Conversely, overexpression of PDK in mouse models maintains low levels of glucose oxidation.
AMPK mediates a longer lasting inhibition of glycolysis by regulating proteins involved in both Glucose and FA oxidation pathways through enzymes such as Glut4, PFK and ACCs. Crucially, during periods of cellular stress, AMPK is able to override the biochemical antagonism of glucose fatty acid cycle to maximally produce ATP to meet energetic requirements.
Randle effect is bidirectional therefore glucose oxidation is also inhibitory to FAO. The production of pyruvate not only inactivates PDKs, but the subsequent production of acetyl-coA which enters TCA produces citrate. Citrate is not completely oxidised and is transported to cytosol where it can reproduce acetyl-coA directed towards FA biosynthesis by generating malonyl-coA. Glucose derived malonyl-coA inhibits the oxidation of FAs, achieved through inhibition of CPT1 and entry of LCFAs into mitochondria. This leads to FAs being directed towards esterification and thus storage. In hepatic cells, long term adapation of glucose results in the upregulation of pyruvate kinase and lipogenic enzymes such as ATP citrate lyase, ACC which is mediated by SREBP and CREBBP transcriptional factors.
Established relationship of glucose-fatty acid cycle in diseases such as diabetes and insulin resistance and other metabolic syndromes. Its role in cancer remains unclear. However metabolic reprogramming is an adaptive feature of most cancers. The ability to remain plastic and utilise different metabolic substrate and storing others for different for imminent use may be provide cancer cells a survival benefit which allows them to adapt to nutrient availability and tumour oxygenation. The metabolic reprogramming evidenced in gliomas are highly complex. Both oxidative and glycolytic phenotypes are features of gliomas. It is possible the reprogramming employed in this cell type may enable flux between metabolic pathways in a spatiotemporal fashion can sustain progression.
Methods of studying tumour progression and cancer cell metabolism in vivo
Magnetic resonance imaging (MRI) and spectroscopy (MRS) are powerful and non-invasive tools used to obtain detailed anatomical information and biochemical signatures in tissue. The combination of imaging and spectroscopy allows the investigation of pathophysiological processes abnormalities associated with disease. 1H MRS is the most commonly used nuclei for in vivo investigations due to the natural abundance of protons in tissue. Spectra can be acquired from regions of interest providing information on metabolism. In vivo brain spectroscopy provides details of the predominantly three tissue components; water, metabolites and macromolecules. Metabolic profiles of normal tissue is relatively steady, however a change in metabolism is an observed characteristic of disease. The metabolites observed frequently in brain spectra include NAA, Cr, Cho, lactate and lipids. NAA is a neuronal marker and decreased levels are often associated with disease state. Cho is a marker associated with increased membrane synthesis which is associated with neoplasms with increased proliferation and expansion. High levels of lipid and lactate has been reported as correlative with poor survival in patients with GBM (REFS needed all over here). Metabolic reprogramming is an established hallmark of cancer including gliomas. It has recently been demonstrated that human derived glioma cells preferentially oxidise fatty acids as their main energy source over glucose (Lin et al. 2016, Mashimo et al. 2014, Maher et al. 2012). Changes in lipid metabolism may therefore prove beneficial in distinguishing normal brain and gliomas.
Aims
The overarching aim of the study is investigate fatty acid oxidation in malignant glioma in syngeneic mouse model. The first arm of the preclinical study is to assess efficacy of Etomoxir (an inihbitior of FAO) in combination with Temozolomide which is the standard chemotherapeutic agent for malignant glioma. The second arm of the study will monitor tumour progression and investigate changes in metabolites that may occur during tumour development.
Methods
Cell culture
Neural stem cells isolated from adult-type C57/B6 mice with an overexpression of Ha-Ras (Ha-RasV12) and HPV18 E6E7 (inhibition of p53 and p16/Rb axis) were maintained serum-free Dulbecco’s modified eagle’s medium/F12 supplemented with 2mM glutamine, 1% N2 and 25ng/ml EGF and FGF.
Syngeneic mouse model of glioma
All in vivo experiments were carried out in accordance with Animals (scientific procedures) Act 1986 (UK Home office) and approved by the local ethics committee (AWERB). Oncogenically transformed neural stem cells isolated from adult-wildtype C57BL/6 mice were transplanted into brain tissue of male adult mice with the same genetic background. A total of 1×104 cells/µl was injected into two sites of striatum, AP +1.0 , ML -1.0, DV -3.5 and -3.0. Animals were monitored daily and clinically scored according to a custom clinical scoresheet. Fourteen days subsequent to intracranial cell implantation, animals were treated with 67mg/kg of Temozolomide or 10%DMSO PBS solution via intraperitoneal injection for 5 days (REFS). Following this, animals were treated daily with 10mg/kg of Etomoxir or olive oil by oral gavage for the duration of the study. Animal drug groups: control n=2 (10%DMSO PBS and olive oil), Etomoxir alone n=1 (10%DMSO PBS and Etomoxir), Temozolomide alone n=2 (TMZ and olive oil) and Combined n=2(10mg/kg ETX and 67mg/kg TMZ). Once treatment commenced, all animals were kept on a soaked diet to ensure feeding whilst receiving daily dosing by gavage. Unfortunately, early loss of animals resulted in low n in drug groups. A contraindication placed during the study was to switch animals that experienced any complications or difficulty to gavaging to coconut pellet which is voluntarily administered (get a photo of this).
In vivo magnetic resonance imaging and spectroscopy
MRI
All Magnetic resonance experiments were acquired using a 7T Varian imaging system equipped with the rapid 33mm volume coil to transmit/receive MR signals. Animals were anaesthetised using a mixture of isoflurane/oxygen gas via facemask. Animals were fitted on a custom loading sled with pneumatic pillow for respiratory and surface temperature monitoring. SA instruments, small animal system was used for physiological monitoring. Scout images were used to ensure animals were correctly orientated for brain imaging. A total of 4 imaging sequences were used to acquire brain images of different contrasts by altering repetition time (TR) and echo time (TE). Parameters for spin echo multi-slice sequence (SEMS) were: SEMS 1 (TR 2000 ms TE 20ms), SEMS 2 (TR 2000 ms TE 20ms with inversion recovery), SEMS 3 (TR 1100 ms TE 60ms) and SEMS 4 (TR 1600 TE 85ms). All image sequences were acquired using matrix 256 x 256, field of view 25 x 25 mm, and slice thickness 1mm. To evaluate tumour development and progression, animals were imaged from day 50 and every 14 days subsequent to that.
1H MR Spectroscopy
Single voxel spectra were acquired from ipsilateral (intracranial injection site) and contralateral (normal brain) for each animal using point resolved spectroscopy (PRESS) sequence. Selection of volume of interest was guided by T1 and T2 weighted images. PRESS sequence parameters: TR 2000ms, TE 8ms, 144ms or 288ms, 4096 real points and 100 averages. Localised manual shimming was performed for each cubic voxel (2mm x 2mm x 2mm) resulting with an unsuppressed water signal with line width between 20-30Hz. Supression of the water signal was optimised using VAPOR ( Variable RF pulse with optimised relaxation delays) water suppression. 1HMR was performed at a short TE of 8ms to acquire signal from a broad range of metabolites. The longer echo-time were used to distinguish the lipid-lactate resonance. At short TEs the methylene (-CH2-) groups of lipids and lactate overlap. However long TEs supress lipids signals due to short relaxation times. At TE 144ms, the resonance of lactate is negative whereas at TE 8ms and TE 288 ms positive in phase- doublet.
Results and data collected
Brain imaging using T1 and T2 weighted imaging sequences
Four separate MR imaging sequences were used to acquire coronal sliced images of the brain with differences in T1 and T2 weighting. Figure shows 1-4 shows images acquired from the different SEMS sequences. The four types of MRI sequences achieve differences in tissue contrast. T1 weighted images Fig. 1-2 have a higher signal to noise but provide limited information on brain structures. For instance, the most noticeable region observed is corpus callosum. By adding an inversion sequence to the T1 weighted image, more distinct brain regions are visualised. The lateral ventricles become more noticeable. The addition of T2weighted sequences resulted in regions of the brain with high CSF being visualised although this image type suffers from a loss of signal and high noise. In combination however, the T1 and T2 images were deemed appropriate for the detection of tumour growth.
Detection of tumour by T1 and T2 weighted MRI
Development of tumour was detected in a control animal in both T1 and T2 weighted images. However, emergence of tumour was detected at earlier at day 78 using T2 weighted MRI compared to T1 weighted. Furthermore, the emergence of MRI visible tumour corresponded with clinical symptoms (e.g. weight loss and tumour associated seizure). At present, no visible tumour has been observed in mice treated with Etomoxir alone, Temozolomide and combination group.

Figure 1. SEMS TR 1000 TE 20
VAriable RF Pulses with
Optimized Relaxation delays
VAriable RF Pulses with
Optimized Relaxation delays

Figure2. SEMS TR 1000 TE 20 with inversion recovery

Figure 3. SEMS TR 1100 TE 60

Figure 4. SEMS TR 1600 TE 85
Example in vivo spectra acquired at short and long TEs
Although in vivo 1HMRS is suffers from high noise to signal ratios, distinct resonances can be observed from residual water at ~4.7 ppm, 3ppm and 1ppm in the spectrum acquired at TE 8ms. Spectral quality at long echo time is reduced due to a higher noise to signal ratio. However at longer echo, residual water signal is not problematic.
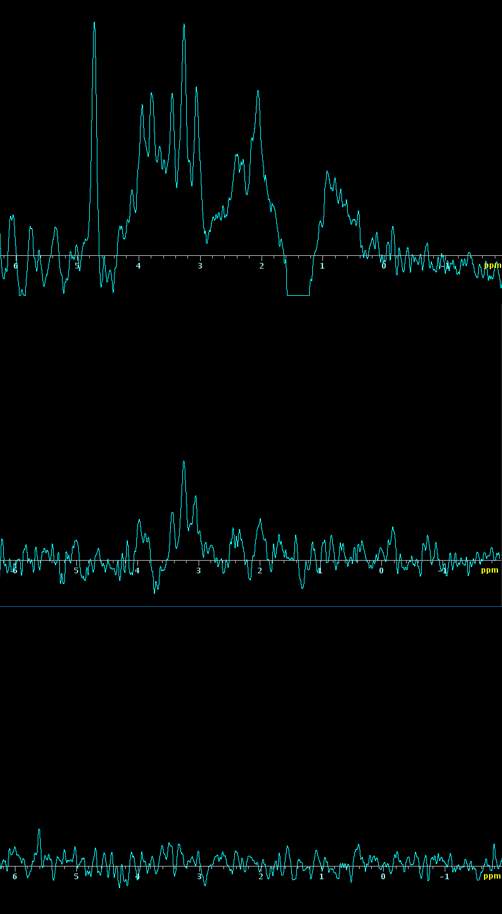
Figure 5. In vivo spectra of PRESS sequence acquired at A) TE 8ms B) 144ms C) 288ms
Metabolic profiles of tumour burdened region vs normal brain
Figure 5 shows water suppressed short TE (8ms) PRESS spectra from 2mm cubic voxels placed on striatum of ipsilateral and contralateral sides of the brain in vivo. The spectra represent 100 averages. A comparison of raw spectra is made impossible due to the residual water signal that causes a distortion of chemical shifts in certain spectral regions. Therefore, post processing is required to accurately fit the data and analysis is required to make a comparative integral measurements of the signal amplitude of metabolite peaks between animals in different treatment groups.
RESULTS
Figure 1a shows a water-suppressed PRESS spectrum from
a2-l voxel placed in the interventricular septum of a
C57Bl/6J mouse in vivo (Fig. 1b) using a quadrature-driven
28 mm birdcage coil; 256 scans were averaged to obtain
this spectrum. The resonances of creatine (methyl- and
methylene-groups), carnitine, taurine, lipids, and the re-
sidual water can be clearly identified. The peaks were
referenced to the residual water signal, set to 4.7 ppm, and
assigned according to Ref. 1
RESULTS
Figure 1a shows a water-suppressed PRESS spectrum from
a2-l voxel placed in the interventricular septum of a
C57Bl/6J mouse in vivo (Fig. 1b) using a quadrature-driven
28 mm birdcage coil; 256 scans were averaged to obtain
this spectrum. The resonances of creatine (methyl- and
methylene-groups), carnitine, taurine, lipids, and the re-
sidual water can be clearly identified. The peaks were
referenced to the residual water signal, set to 4.7 ppm, and
assigned according to Ref. 1
RESULTS
Figure 1a shows a water-suppressed PRESS spectrum from
a2-l voxel placed in the interventricular septum of a
C57Bl/6J mouse in vivo (Fig. 1b) using a quadrature-driven
28 mm birdcage coil; 256 scans were averaged to obtain
this spectrum. The resonances of creatine (methyl- and
methylene-groups), carnitine, taurine, lipids, and the re-
sidual water can be clearly identified. The peaks were
referenced to the residual water signal, set to 4.7 ppm, and
assigned according to Ref. 1

Figure 6 In vivo spectra of Short TE acquired from ipsilateral and contralateral hemispheres.
Conclusion
Over the course of the study, I have:
I have successfully tracked tumour progression using both T1 and T2 weighted MR imaging protocols in a syngeneic mouse model of glioma. Additionally, I have acquired in vivo spectroscopy data during tumour progression from two distinct brain regions (including the control hemisphere, where tumour cells were not implanted). As the study remains ongoing, a comparison of tumour development and metabolite signatures between drug treatment groups is yet to be made.
Future directions of the study
MRI data analysis
Histological analysis.
Spectroscopy
In vivo efficacy study
The next considerable aim of the project will be to carry out a double blinded preclinical study determining efficacy of Etomoxir in combination with Temozolomide for slowing progression of glioma.
In vitro
In vitro NMRSof metabolic profile of mouse NSCs and human HGBMs treated with appropriate drugs.
Identify changes in lipid droplets with each of the drug treatment conditions.
Identify changes in proliferation and viability with each of the drug treatment condition.
List of references
Bonnet, S., Archer, S.L., Allalunis-Turner, J., Haromy, A., Beaulieu, C., Thompson, R., Lee, C.T., Lopaschuk, G.D., Puttagunta, L., Harry, G., Hashimoto, K., Porter, C.J., Andrade, M.A., Thebaud, B. and Michelakis, E.D. (2007) ‘A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth’, Cancer Cell, 11(1), pp. 37-51.
Brennan, C.W., Verhaak, R.G.W., McKenna, A., Campos, B., Noushmehr, H., Salama, S.R., Zheng, S.Y., Chakravarty, D., Sanborn, J.Z., Berman, S.H., Beroukhim, R., Bernard, B., Wu, C.J., Genovese, G., Shmulevich, I., Barnholtz-Sloan, J., Zou, L.H., Vegesna, R., Shukla, S.A., Ciriello, G., Yung, W.K., Zhang, W., Sougnez, C., Mikkelsen, T., Aldape, K., Bigner, D.D., Van Meir, E.G., Prados, M., Sloan, A., Black, K.L., Eschbacher, J., Finocchiaro, G., Friedman, W., Andrews, D.W., Guha, A., Iacocca, M., O’Neill, B.P., Foltz, G., Myers, J., Weisenberger, D.J., Penny, R., Kucherlapati, R., Perou, C.M., Hayes, D.N., Gibbs, R., Marra, M., Mills, G.B., Lander, E., Spellman, P., Wilson, R., Sander, C., Weinstein, J., Meyerson, M., Gabriel, S., Laird, P.W., Haussler, D., Getz, G., Chin, L. and Network, T.R. (2014) ‘The Somatic Genomic Landscape of Glioblastoma (Vol 155, pg 462, 2013)’, Cell, 157(3), pp. 753-753.
Cabodevilla, A.G., Sanchez-Caballero, L., Nintou, E., Boiadjieva, V.G., Picatoste, F., Gubern, A. and Claro, E. (2013) ‘Cell Survival during Complete Nutrient Deprivation Depends on Lipid Droplet-fueled beta-Oxidation of Fatty Acids’, Journal of Biological Chemistry, 288(39), pp. 27777-27788.
Chen, W., Cloughesy, T., Kamdar, N., Satyamurthy, N., Bergsneider, M., Liau, L., Mischel, P., Czernin, J., Phelps, M.E. and Silverman, D.H.S. (2005) ‘Imaging proliferation in brain tumors with F-18-FLT PET: Comparison with F-18-FDG’, Journal of Nuclear Medicine, 46(6), pp. 945-952.
DeBerardinis, R.J., Lum, J.J., Hatzivassiliou, G. and Thompson, C.B. (2008) ‘The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation’, Cell Metabolism, 7(1), pp. 11-20.
Diedrich, M. and Henschel, K.P. (1990) ‘THE NATURAL OCCURRENCE OF UNUSUAL FATTY-ACIDS .1. ODD NUMBERED FATTY-ACIDS’, Nahrung-Food, 34(10), pp. 935-943.
Ensenauer, R., He, M., Willard, J.M., Goetzman, E.S., Corydon, T.J., Vandahl, B.B., Mohsen, A.W., Isaya, G. and Vockley, J. (2005) ‘Human acyl-CoA dehydrogenase-9 plays a novel role in the mitochondrial beta-oxidation of unsaturated fatty acids’, Journal of Biological Chemistry, 280(37), pp. 32309-32316.
Geng, F., Cheng, X., Wu, X.N., Cheng, C.M., Guo, J.Y., Horbinski, C., Kaur, B., Chakravarti, A. and Guo, D.L. (2016) ‘LIPID DROPLETS, A NOVEL DIAGNOSTIC BIOMARKER AND METABOLIC TARGET IN GLIOBLASTOMA’, Neuro-Oncology, 18, pp. 36-36.
Gimeno, R.E. (2007) ‘Fatty acid transport proteins’, Current Opinion in Lipidology, 18(3), pp. 271-276.
Grube, S., Dunisch, P., Freitag, D., Klausnitzer, M., Sakr, Y., Walter, J., Kalff, R. and Ewald, C. (2014) ‘Overexpression of fatty acid synthase in human gliomas correlates with the WHO tumor grade and inhibition with Orlistat reduces cell viability and triggers apoptosis’, Journal of Neuro-Oncology, 118(2), pp. 277-287.
Guppy, M., Leedman, P., Zu, X.L. and Russell, V. (2002) ‘Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells’, Biochemical Journal, 364, pp. 309-315.
Hanahan, D. and Weinberg, R.A. (2000) ‘The hallmarks of cancer’, Cell, 100(1), pp. 57-70.
Hanahan, D. and Weinberg, R.A. (2011) ‘Hallmarks of Cancer: The Next Generation’, Cell, 144(5), pp. 646-674.
Hardie, D.G. and Alessi, D.R. (2013) ‘LKB1 and AMPK and the cancer-metabolism link – ten years after’, Bmc Biology, 11.
Hardie, D.G. and Pan, D.A. (2002) ‘Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase’, Biochemical Society Transactions, 30, pp. 1064-1070.
Hardie, D.G., Ross, F.A. and Hawley, S.A. (2012) ‘AMPK: a nutrient and energy sensor that maintains energy homeostasis’, Nature Reviews Molecular Cell Biology, 13(4), pp. 251-262.
Hegi, M.E., Diserens, A., Gorlia, T., Hamou, M., de Tribolet, N., Weller, M., Kros, J.M., Hainfellner, J.A., Mason, W., Mariani, L., Bromberg, J.E.C., Hau, P., Mirimanoff, R.O., Cairncross, J.G., Janzer, R.C. and Stupp, R. (2005) ‘MGMT gene silencing and benefit from temozolomide in glioblastoma’, New England Journal of Medicine, 352(10), pp. 997-1003.
Heiden, M.G.V., Cantley, L.C. and Thompson, C.B. (2009) ‘Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation’, Science, 324(5930), pp. 1029-1033.
Herms, A., Bosch, M., Reddy, B.J.N., Schieber, N.L., Fajardo, A., Ruperez, C., Fernandez-Vidal, A., Ferguson, C., Rentero, C., Tebar, F., Enrich, C., Parton, R.G., Gross, S.P. and Pol, A. (2015) ‘AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation’, Nature Communications, 6.
Houten, S.M. and Wanders, R.J.A. (2010) ‘A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation’, Journal of Inherited Metabolic Disease, 33(5), pp. 469-477.
Lee, J. and Wolfgang, M.J. (2012) ‘Metabolomic profiling reveals a role for CPT1c in neuronal oxidative metabolism’, Bmc Biochemistry, 13.
Lin, H., Patel, S., Affleck, V., Wilson, I., Turnbull, D., Joshi, A., Maxwell, R. and Stoll, E. (2016) ‘FATTY ACID OXIDATION IS REQUIRED FOR THE RESPIRATION AND PROLIFERATION OF MALIGNANT GLIOMA CELLS’, Neuro-Oncology, 18, pp. 36-36.
Louis, D.N., Ohgaki, H., Wiestler, O.D., Cavenee, W.K., Burger, P.C., Jouvet, A., Scheithauer, B.W. and Kleihues, P. (2007) ‘The 2007 WHO classification of tumours of the central nervous system’, Acta Neuropathologica, 114(2), pp. 97-109.
Louis, D.N., Perry, A., Reifenberger, G., von Deimling, A., Figarella-Branger, D., Cavenee, W.K., Ohgaki, H., Wiestler, O.D., Kleihues, P. and Ellison, D.W. (2016) ‘The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary’, Acta Neuropathologica, 131(6), pp. 803-820.
Malmer, B., Adatto, P., Armstrong, G., Barnholtz-Sloan, J., Bernstein, J.L., Claus, E., Davis, F., Houlston, R., Il’yasova, D., Jenkins, R., Johansen, C., Lai, R., Lau, C., McCarthy, B., Nielsen, H., Olson, S.H., Sadetzki, S., Shete, S., Wiklund, F., Wrensch, M., Yang, P. and Bondy, M. (2007) ‘GLIOGENE – an international consortium to understand familial glioma’, Cancer Epidemiology Biomarkers & Prevention, 16(9), pp. 1730-1734.
Mashimo, T., Pichumani, K., Vemireddy, V., Hatanpaa, K.J., Singh, D.K., Sirasanagandla, S., Nannepaga, S., Piccirillo, S.G., Kovacs, Z., Foong, C., Huang, Z.G., Barnett, S., Mickey, B.E., DeBerardinis, R.J., Tu, B.P., Maher, E.A. and Bachoo, R.M. (2014) ‘Acetate Is a Bioenergetic Substrate for Human Glioblastoma and Brain Metastases’, Cell, 159(7), pp. 1603-1614.
McGarry, J.D., Leatherman, G.F. and Foster, D.W. (1978) ‘CARNITINE PALMITOYLTRANSFERASE I – SITE OF INHIBITION OF HEPATIC FATTY-ACID OXIDATION BY MALONYL-COA’, Journal of Biological Chemistry, 253(12), pp. 4128-4136.
Menendez, J.A. and Lupu, R. (2007) ‘Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis’, Nature Reviews Cancer, 7(10), pp. 763-777.
Michelakis, E.D., Sutendra, G., Dromparis, P., Webster, L., Haromy, A., Niven, E., Maguire, C., Gammer, T.L., Mackey, J.R., Fulton, D., Abdulkarim, B., McMurtry, M.S. and Petruk, K.C. (2010) ‘Metabolic Modulation of Glioblastoma with Dichloroacetate’, Science Translational Medicine, 2(31).
Ostrom, Q.T., Gittleman, H., Xu, J., Kromer, C., Wolinsky, Y., Kruchko, C. and Barnholtz-Sloan, J.S. (2016) ‘CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009-2013’, Neuro-Oncology, 18, pp. v1-v75.
Oudard, S., Arvelo, F., Miccoli, L., Apiou, F., Dutrillaux, A.M., Poisson, M., Dutrillaux, B. and Poupon, M.F. (1996) ‘High glycolysis in gliomas despite low hexokinase transcription and activity correlated to chromosome 10 loss’, British Journal of Cancer, 74(6), pp. 839-845.
Park, J.H., Vithayathil, S., Kumar, S., Sung, P.L., Dobrolecki, L.E., Putluri, V., Bhat, V.B., Bhowmik, S.K., Gupta, V., Arora, K., Wu, D.L., Tsouko, E., Zhang, Y.Q., Maity, S., Donti, T.R., Graham, B.H., Frigo, D.E., Coarfa, C., Yotnda, P., Putluri, N., Sreekumar, A., Lewis, M.T., Creighton, C.J., Wong, L.J.C. and Kaipparettu, B.A. (2016) ‘Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer’, Cell Reports, 14(9), pp. 2154-2165.
Phillips, H.S., Kharbanda, S., Chen, R.H., Forrest, W.F., Soriano, R.H., Wu, T.D., Misra, A., Nigro, J.M., Colman, H., Soroceanu, L., Williams, P.M., Modrusan, Z., Feuerstein, B.G. and Aldape, K. (2006) ‘Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis’, Cancer Cell, 9(3), pp. 157-173.
Pike, L.S., Smift, A.L., Croteau, N.J., Ferrick, D.A. and Wu, M. (2011) ‘Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells’, Biochimica Et Biophysica Acta-Bioenergetics, 1807(6), pp. 726-734.
Rajneesh, K.F. and Binder, D.K. (2009) ‘Tumor-associated epilepsy’, Neurosurgical Focus, 27(2).
Reilly, P.T. and Mak, T.W. (2012) ‘Molecular Pathways: Tumor Cells Co-opt the Brain-Specific Metabolism Gene CPT1C to Promote Survival’, Clinical Cancer Research, 18(21), pp. 5850-5855.
Ricciardi, M.R., Mirabilii, S., Allegretti, M., Licchetta, R., Calarco, A., Torrisi, M.R., Foa, R., Nicolai, R., Peluso, G. and Tafuri, A. (2015) ‘Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias’, Blood, 126(16), pp. 1925-1929.
Rohrig, F. and Schulze, A. (2016) ‘The multifaceted roles of fatty acid synthesis in cancer’, Nature Reviews Cancer, 16(11), pp. 732-749.
Samudio, I., Harmancey, R., Fiegl, M., Kantarjian, H., Konopleva, M., Korchin, B., Kaluarachchi, K., Bornmann, W., Duvvuri, S., Taegtmeyer, H. and Andreeff, M. (2010) ‘Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction’, Journal of Clinical Investigation, 120(1), pp. 142-156.
Schlaepfer, I.R., Rider, L., Rodrigues, L.U., Gijon, M.A., Pac, C.T., Romero, L., Cimic, A., Sirintrapun, S.J., Glode, L.M., Eckel, R.H. and Cramer, S.D. (2014) ‘Lipid Catabolism via CPT1 as a Therapeutic Target for Prostate Cancer’, Molecular Cancer Therapeutics, 13(10), pp. 2361-2371.
Stupp, R., Mason, W.P., van den Bent, M.J., Weller, M., Fisher, B., Taphoorn, M.J.B., Belanger, K., Brandes, A.A., Marosi, C., Bogdahn, U., Curschmann, J., Janzer, R.C., Ludwin, S.K., Gorlia, T., Allgeier, A., Lacombe, D., Cairncross, J.G., Eisenhauer, E., Mirimanoff, R.O., Van Den Weyngaert, D., Kaendler, S., Krauseneck, P., Vinolas, N., Villa, S., Wurm, R.E., Maillot, M.H.B., Spagnolli, F., Kantor, G., Malhaire, J.P., Renard, L., De Witte, O., Scandolaro, L., Vecht, C.J., Maingon, P., Lutterbach, J., Kobierska, A., Bolla, M., Souchon, R., Mitine, C., Tzuk-Shina, T., Kuten, A., Haferkamp, G., de Greve, J., Priou, F., Menten, J., Rutten, I., Clavere, P., Malmstrom, A., Jancar, B., Newlands, E., Pigott, K., Twijnstra, A., Chinot, O., Reni, M., Boiardi, A., Fabbro, M., Campone, M., Bozzino, J., Frenay, M., Gijtenbeek, J., Delattre, J.Y., De Paula, U., Hanzen, C., Pavanato, G., Schraub, S., Pfeffer, R., Soffietti, R., Kortmann, R.D., Taphoorn, M., Torrecilla, J.L., Grisold, W., Huget, P., Forsyth, P., Fulton, D., Kirby, S., Wong, R., Fenton, D., Cairncross, G., Whitlock, P., Burdette-Radoux, S., Gertler, S., Saunders, S., Laing, K., Siddiqui, J., Martin, L.A., Gulavita, S., Perry, J., Mason, W., Thiessen, B., Pai, H., Alam, Z.Y., Eisenstat, D., Mingrone, W., Hofer, S., Pesce, G., Dietrich, P.Y., Thum, P., Baumert, B., Ryan, G. and European Org Res Treatment Canc, B. (2005) ‘Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma’, New England Journal of Medicine, 352(10), pp. 987-996.
Tao, B.B., He, H., Shi, X.H., Wang, C.L., Li, W.Q., Li, B., Dong, Y., Hu, G.H., Hou, L.J., Luo, C., Chen, J.X., Chen, H.R., Yu, Y.H., Sun, Q.F. and Lu, Y.C. (2013) ‘Up-regulation of USP2a and FASN in gliomas correlates strongly with glioma grade’, Journal of Clinical Neuroscience, 20(5), pp. 717-720.
Verhaak, R.G.W., Hoadley, K.A., Purdom, E., Wang, V., Qi, Y., Wilkerson, M.D., Miller, C.R., Ding, L., Golub, T., Mesirov, J.P., Alexe, G., Lawrence, M., O’Kelly, M., Tamayo, P., Weir, B.A., Gabriel, S., Winckler, W., Gupta, S., Jakkula, L., Feiler, H.S., Hodgson, J.G., James, C.D., Sarkaria, J.N., Brennan, C., Kahn, A., Spellman, P.T., Wilson, R.K., Speed, T.P., Gray, J.W., Meyerson, M., Getz, G., Perou, C.M., Hayes, D.N. and Canc Genome Atlas Res, N. (2010) ‘Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1’, Cancer Cell, 17(1), pp. 98-110.
Vlashi, E., Lagadec, C., Vergnes, L., Matsutani, T., Masui, K., Poulou, M., Popescu, R., Della Donna, L., Evers, P., Dekmezian, C., Reue, K., Christofk, H., Mischel, P.S. and Pajonk, F. (2011) ‘Metabolic state of glioma stem cells and nontumorigenic cells’, Proceedings of the National Academy of Sciences of the United States of America, 108(38), pp. 16062-16067.
Welte, M.A. (2015) ‘Expanding Roles for Lipid Droplets’, Current Biology, 25(11), pp. R470-R481.
Wolfgang, M.J., Kurama, T., Dai, Y., Suwa, A., Asaumi, M., Matsumoto, S., Cha, S.H., Shimokawa, T. and Lane, M.D. (2006) ‘The brain-specific carnitine palmitoyltransferase-1c regulates energy homeostasis’, Proceedings of the National Academy of Sciences of the United States of America, 103(19), pp. 7282-7287.
Zaugg, K., Yao, Y., Reilly, P.T., Kannan, K., Kiarash, R., Mason, J., Huang, P., Sawyer, S.K., Fuerth, B., Faubert, B., Kalliomaki, T., Elia, A., Luo, X.Y., Nadeem, V., Bungard, D., Yalavarthi, S., Growney, J.D., Wakeham, A., Moolani, Y., Silvester, J., Ten, A.Y., Bakker, W., Tsuchihara, K., Berger, S.L., Hill, R.P., Jones, R.G., Tsao, M., Robinson, M.O., Thompson, C.B., Pan, G.H. and Mak, T.W. (2011) ‘Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress’, Genes & Development, 25(10), pp. 1041-1051.
Zhang, J., Zhang, W.P., Zou, D.J., Chen, G.Y., Wan, T., Zhang, M.H. and Cao, X.T. (2002) ‘Cloning and functional characterization of ACAD-9, a novel member of human acyl-CoA dehydrogenase family’, Biochemical and Biophysical Research Communications, 297(4), pp. 1033-1042.
Zhou, Y.F., Zhou, Y., Shingu, T., Feng, L., Chen, Z., Ogasawara, M., Keating, M.J., Kondo, S. and Huang, P. (2011) ‘Metabolic Alterations in Highly Tumorigenic Glioblastoma Cells PREFERENCE FOR HYPOXIA AND HIGH DEPENDENCY ON GLYCOLYSIS’, Journal of Biological Chemistry, 286(37), pp. 32843-32853.
You have to be 100% sure of the quality of your product to give a money-back guarantee. This describes us perfectly. Make sure that this guarantee is totally transparent.
Read moreEach paper is composed from scratch, according to your instructions. It is then checked by our plagiarism-detection software. There is no gap where plagiarism could squeeze in.
Read moreThanks to our free revisions, there is no way for you to be unsatisfied. We will work on your paper until you are completely happy with the result.
Read moreYour email is safe, as we store it according to international data protection rules. Your bank details are secure, as we use only reliable payment systems.
Read moreBy sending us your money, you buy the service we provide. Check out our terms and conditions if you prefer business talks to be laid out in official language.
Read more